
Abstract
The study describes the transmission of a CTX-M-15-producing ST15 Klebsiella pneumoniae between patients treated in a single center and the subsequent inter-institutional spread by patient referral occurring between May 2012 and September 2013. A suspected epidemiological link between clinical K. pneumoniae isolates was supported by patient contact tracing and genomic phylogenetic analysis from May to November 2012. By May 2013, a patient treated in three institutions in two cities was involved in an expanding cluster caused by this high-risk clone (HiRiC) (local expansion, CTX-M-15 producing, and containing hypervirulence factors). A clone-specific multiplex PCR was developed for patient screening by which another patient was identified in September 2013. Genomic phylogenetic analysis including published ST15 genomes revealed a close homology with isolates previously found in the USA. Environmental contamination and lack of consistent patient screening were identified as being responsible for the clone dissemination. The investigation addresses the advantages of whole-genome sequencing in the early detection of HiRiC with a high propensity of nosocomial transmission and prolonged circulation in the regional patient population. Our study suggests the necessity for inter-institutional/regional collaboration for infection/outbreak management of K. pneumoniae HiRiCs.
Similar content being viewed by others

Introduction
Klebsiella pneumoniae has emerged as an important nosocomial pathogen, known as one of the “ESKAPE” pathogens1. Especially, the prevalence of multi-drug resistant (MDR) K. pneumoniae increased dramatically in recent years. This limits efficient clinical treatment tremendously resulting in undesirable treatment outcomes.
To control the spread of drug resistance, the tracking of antimicrobial resistant microorganisms is crucial and has been proposed in the CDC antimicrobial resistance action plan as one of the four core actions2. Use of multilocus sequence typing (MLST) revealed that the population of MDR K. pneumoniae is largely oligoclonal, and some epidemic/endemic clones have been identified. For instance, the extended-spectrum β-lactamase (ESBL)-producing K. pneumoniae (ESBL-KP) mainly belong to sequence type (ST) 11, ST15, ST101, ST147, and ST3363,4,5,6,7, and a specific lineage (ST258) played a major role in dissemination of Klebsiella pneumoniae carbapenemases (KPC) worldwide8. With the rapid development of whole-genome sequencing (WGS), the population structure of MDR pathogens can be dissected at a higher resolution level. This challenges conventional hypotheses on clonal descent of K. pneumoniae.
To efficiently control and prevent the dissemination of MDR pathogens in hospital settings, the ability of early detection and the tracking of infected or colonized patients are crucial9. This is especially a challenge during a prolonged outbreak, as transmissions between patients may be caused by additional reservoirs (e.g. health-care workers and the environment), and/or in case different institutes and/or cities are involved in the outbreak.
In this study, we present our experience in using whole-genome sequencing and further developing an outbreak clone-specific PCR for tracing and controlling a regional and inter-institutional outbreak caused by a ST15 CTX-M-15-KP clone between May 2012 and September 2013 in the northern part of Netherlands.
Materials and Methods
Setting
The University Medical Center Groningen (UMCG) is a 1,300-bed tertiary-care medical centre in the northern part of Netherlands. The rehabilitation centre is part of the university hospital but located 6-kilometer away from the main centre. Most patients of the rehabilitation center were transferred from the university hospital. If a complication occurred or if additional treatment was required, they were admitted to or visiting the hospital again. Part of the medical staff worked both in the hospital and the rehabilitation center.
Patient tracking and contact tracing
At the end of August 2012, an increase of ESBL-producing K. pneumoniae (ESBL-KP) was observed at the UMCG and the associated rehabilitation centre. To prevent further spread, stringent infection control measures consisting of strict patient and staff cohorting were introduced at both sites in August 2012. The outbreak was declared under control in September 2012. Extended infection control measures ended in May 2013. Patients on “high risk” wards, defined as those wards having extensive patient exchange with the ones where positive patients stayed, were screened for ESBL-KP once a week. Patients overlapping with positive patients on the ward were screened twice a week. Discharged patients who overlapped with positive patients between May and September 2012 were traced and retrospectively screened at home using a self-sampling kit. The kit contained two swabs used for the throat and rectum sampling. Instructions about how to use the kit were included. All patients were requested to send the kit back as soon as they took the samples. A case was defined as a patient infected or colonized with an ESBL-producing K. pneumoniae after 1st May 2012 sharing the same antibiotic resistance pattern as the outbreak clone.
Screening and isolates collected in this study
Screening of ESBL-producing Enterobacteriaceae (ESBL-E) was performed as described previously10. Briefly, patients were screened for ESBL-E carriage using perianal swabs (Eswab, Copan, Italy), and environmental screening was performed in the patient rooms (e.g. beds) and toilets (e.g. toilet chairs) using MW728 POLYWIPE® sponge swabs (Medical wire & equipment, Wiltshire, England). The swab was plated on a blood agar plate after vortexing, and the liquid Amies eluent was inoculated on selective tryptic soy broth with cefotaxime (0.25 mg/L) and vancomycin (8 mg/L) (TSB-VC). After 18–24 hours of incubation (35–37 °C), 10 μl TSB-VC was sub-cultured on both sides of an Extended Beta-Lactamase Screening Agar (EbSA) plate (AlphaOmega, ‘s-Gravenhage, Netherlands). The EbSA plate consists of a split MacConkey agar plate containing ceftazidime (1.0 mg/L) on one side and cefotaxime (1.0 mg/L) on the other side. Both sides contain cloxacillin (400 mg/L) and vancomycin (64 mg/L) for inhibition of AmpC beta-lactamase-producing bacteria and Gram-positive bacteria, respectively. Subsequently the plates were incubated aerobically at 35 to 37 °C for 18 to 24 hours. Species identification was performed for all oxidase negative isolates that grew on either side of the agar by MALDI-TOF (Bruker Daltonik GmbH, Bremen, Germany). In total, 19 K. pneumoniae isolates obtained from patients (KP-1 to KP12, KP-45D, KP-33P, KP-86L, and KP-33F) and environment (KP-1E to KP-3E) were included in this study. The details of isolates are listed in Table 1.
Conventional testing
Phenotypic susceptibility testing was performed using the Vitek II system with card AST N-199 (BioMerieux, Marcy l′Etoile, France) according to the guidelines of the manufacturer. Breakpoints were interpreted according to EUCAST guidelines (bacteria v4.0). MLST was performed according to the protocol described on the K. pneumoniae MLST website (www.pasteur.fr/mlst). The sequence type (ST) was assigned by the MLST database (www.pasteur.fr/mlst/Kpneumoniae.html).
WGS, de novo assembly and annotation
A total of 19 isolates were sequenced. WGS and de novo assembly was performed as described previously11. Isolate KP-5 was randomly selected for being scaffolded by mate-pair sequencing. The mate-pair DNA library was prepared using the Mate Pair Library Prep Kit v2 (Illumina) according to the manufacturer’s instructions followed by running on the Miseq for generating 100-bp reads. Scaffolding was performed by SSPACE standard version 3.0 with default settings12. Further gaps within scaffolds were attempted to be closed using GapFiller with default settings13. The genomes were manually curated after performing automatic annotation on the RAST server14.
Identification of antibiotic resistance-related genes and virulence factors
Three types of drug-resistance genes were investigated, including acquired drug-resistance genes, genes of efflux pumps, and genes associated with drug resistance by specific mutations. Virulence factors studied here include adhesins, capsule production, iron uptake systems, nitrogen source utilization and secretion systems15,16. The acquired antimicrobial resistance genes were identified by uploading assembled genomes to the Resfinder server v2.1 (http://cge.cbs.dtu.dk/services/ResFinder-2.1/). The other genes relating to resistance and virulence were detected by using the mapping unit of CLC Genomics Workbench to map and/or by blasting assembled genomes to a pseudomolecule generated by concatenating a set of target genes. The capsular genotype was determined in silico by wzi typing17. Scaffolds with resistance-related and virulence genes were blasted against GenBank to identify their genetic location.
Detection of single-nucleotide polymorphisms (SNPs)
The scaffolded genome of isolate KP-5 was ordered and oriented relative to the finished genome K. pneumoniae PMK1 (Accession number: CP008929) via ABACAS18. Reads of other isolates were mapped to the rearranged KP-5 genome by CLC Genomics Workbench with default settings. Candidate SNPs were detected by the algorithm “Quality-based variant detection” of CLC Genomics Workbench. SNPs were filtered out as described previously to acquire high-quality core-genome SNPs9.
Core-genome phylogenetic analysis
Fragments (≥500 bp) shared by all genomes were collected and then concatenated resulting in a pseudomolecule defined as the core genome. The detected high-quality SNPs from outbreak isolates were used for SNP-based phylogenetic reconstruction by RAxML v7.4.219 with 1000 bootstrap replications under the general time-reversible model with Gamma correction (GTR+G). Assembled genomes and genomes retrieved from GenBank were aligned by ProgressiveMauve20. The alignment of core genomes was used for estimating the maximum likelihood (ML) phylogeny by RAxML v7.4.2 as mentioned above.
Identification of DNA signatures and development of outbreak-specific multiplex PCR
The core genome of outbreak isolates was blasted against our local K. pneumoniae genome database (76 genomes with diverse sequence types). The unique fragments were extracted and blasted against GenBank (update to 20 October 2014). The non-match fragments not related to mobile genetic elements (e.g. phages, plasmids, and transposons) were considered as the DNA signatures for the outbreak clone. Primers for multiplex PCR specific to DNA signatures were designed by MPprimer21. All PCR-positive isolates were confirmed and further characterized by WGS.
Results
Outbreak description and intervention
The suspected index patient (patient 1) had been admitted to a hospital in Gambia for necrotizing pancreatitis before arriving to the Netherlands and was referred from a regional acute care hospital to the UMCG in May 2012. As an MRSA-screening test was negative before the patient arrived at the UMCG patient 1 was not placed in isolation upon admission. Subsequently, the patient was screened for ESBL-producing Enterobacteriaceae and tested positive for ESBL-KP. The patient therefore was immediately put into contact isolation which implied that medical staff was required to wear a gown and gloves when having contact with the patient. A second patient (patient 2) stayed three days at the same ward with patient 1 and was later found positive for an ESBL-KP during the ICU stay. Two further patients were identified (patient 3 and 4) staying on the same ICU as patient 2. The isolates obtained from these patients had identical antibiotic resistance features as those of patient 1 and 2. This triggered an investigation consisting of patient tracking, contact tracing and screening. Subsequently, another five patients (patient 5 to 9) sharing the same ward and facilities with patient 3 at the rehabilitation centre were found to be positive for ESBL-KP. Supplementary Fig. S1 illustrates the intra-hospital patient transfer. In November 2012, an ESBL-KP was isolated from another patient (patient 10) staying at the same ward as patients 3 and 8 who were treated in contact isolation.
In May 2013, a patient (patient 11) admitted to the neurological ICU at the UMCG was found to carry an ESBL-KP phenotypically indistinguishable from the 2012 outbreak strain on admission screening. Based on the genomic similarity between the isolate of patient 11 and the 2012 outbreak isolates, we were able to predict the grade of epidemiological relatedness (see below). Patient 11 had no contact with any of the ten positive patients. Other patients at the neurological ICU were repeatedly screened and tested negative. Patient 11 was transferred from a secondary hospital in a city about 80-kilometer south of the UMCG. Further investigation revealed that patient 11 resided in the room where patient 6, still positive for ESBL-KP, had stayed after referral from the rehabilitation centre to the secondary acute care hospital. In fact, a small outbreak with 4 patients was reported but isolates were not available for this investigation.
In September 2013, a patient (patient 12) admitted at the UMCG was identified to carry an ESBL-KP highly similar to the 2012 outbreak isolates using a clone-specific PCR (see below). No positive patients were found at the hospital during that time. Epidemiological investigation revealed that before this admission patient 12 resided in a room in November 2012 where patient 1 stayed two months before.
In total, twelve patients positive for a phylogenetically highly related strain (see below) were identified by screening more than 120 patients from May 2012 until September 2013. Seven of them developed a clinical infection (Table 1). Figure 1 shows the most likely transmission route between the patients.

The transmission route was reconstructed by epidemiological and genomic data (see Appendix Materials and Methods for details of reconstruction methods). Each node represents a patient, and an arrow indicates a possible transmission event from one patient to another. The blue arrow with solid line represents a direct transmission event supported by both epidemiological data and genetic data, the blue arrow with dash line represents an indirect transmission (e.g. via environment) supported by epidemiological data, and the red arrow indicates the equally parsimonious transmission link which cannot be resolved by neither epidemiological data nor genetic data. The inter-institutional transfer of the patient is shown by dash line, on which the distance between institutions is indicated. The red star represents an outbreak at the secondary hospital, but the isolates were unavailable for our investigation.
Antimicrobial susceptibility and MLST typing
The 12 isolates (KPO-1 to KPO-12) from the 12 positive patients shared identical antibiotic resistance patterns. They were ESBL positive and resistant to amoxicillin, amoxicillin-clavulanic-acid, cefuroxime, cefotaxime, ceftazidime, gentamicin, tobramycin, co-trimoxazole, ciprofloxacin, cefepime, norfloxacin, and trimethoprim.
The MLST typing results showed that all isolates were assigned to ST15 (MLST profile: 1-1-1-1-1-1-1), which is known as the major member of the epidemic clonal group (CG) 153.
Population genomic analysis
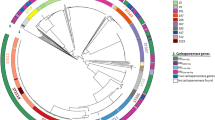
Phylogenetic analysis of the core genome carried out for isolates from patients (1–4) early during the course of the investigation revealed their isogenicity. Additional isolates from patients (5–10) identified through contact tracing and screening as well as isolate KP-11 from sporadic patient 11 in 2013 all fell into the same genetic cluster (Fig. 2). The 11 isolates differed by only 7 SNPs in inter- and intragenic regions across the core genome (see Supplementary Table S1). Indeed, three isolates obtained from patients at the UMCG (KP-1, KP-2 and KP-4) and another two from patients of the rehabilitation centre (KP-6 and KP-7) were identical, i.e. they had no SNPs in the core genome. This phylogeny is concordant with available epidemiological data. We also predicted that the 2013 isolate KP-11 was derived from the outbreak clone, and differed by 2-4 SNPs from available 2012 outbreak isolates (Fig. 2).

A maximum likelihood tree was constructed based on the alignments of a 4.47 Mb genome, defined as the core genome in this study. The tree was mid-point rooted. The size of node represents the percentage of bootstrap support, and the biggest one is equal to 100. Sequence types are indicated between brackets. The isolation time and resource of all ST15 strains are shown. The cluster of outbreak isolates is simplified as a red triangle. The non-outbreak isolates sequenced in this study are in blue, and the others retrieved from GenBank are in black. The inset shows the close-up unrooted tree of outbreak isolates, in which the patient isolates are shown in red (2012) and purple (2013), and the environment isolates are in green. The number of SNPs is indicated on the branches.
To assess the population diversity of K. pneumoniae ST15, four non-outbreak ESBL-KP isolates obtained before and after the outbreak period at the UMCG (Table 1), and 10 strains retrieved from GenBank (see Supplementary Table S2 online) were included in the phylogenetic analysis. The ST15 strains were split into two distinct clades (I and II). Five ST15 strains (MGH63, MGH65, MGH75, BAMC07-18 and BIDMC-33B) isolated in US clustered with the outbreak clone (clade I) (Fig. 2). A non-outbreak ST15 strain KP-33P isolated from a patient at the UMCG in 2010 was separated from the outbreak clone, but clustered tightly with an ST15 NDM-1-producing isolate PMK1 (82 SNPs) (clade II) causing a nosocomial outbreak in Nepal 201222.Another non-outbreak ST437 isolate KP-86L clustered with isolates of ST11 and ST258, belonging to the epidemic clone group 258.
Patient screening by an outbreak-specific multiplex PCR
Three DNA signatures specific to the outbreak clone were identified. Two of them consisted of coding sequences for a hypothetical protein. The other one was located within the capsule biosynthesis region, which consisted of a glycosyltransferase-encoding gene. Primers for multiplex PCR were designed based on these three signatures (see Supplementary Table S3 online). To assess the discriminatory capability of the primers, we tested 35 isolates representing at least 20 different STs including ST15 (one isolate). The result showed that only the outbreak isolates were positive in the multiplex PCR, and that non-outbreak ST15 isolates can be successfully discriminated from the outbreak isolates (see Supplementary Fig. S2).
In September 2013, an isolate KP-12 sharing an identical antibiogram with the outbreak clone was obtained from patient 12 (Table 1), and was identified to belong to the outbreak clone as the outbreak-specific PCR was positive. In March 2015, the outbreak-specific PCR was applied to exclude the genetic relation of three isolates obtained from 3 patients staying at the same ward, and having a similar antibiogram as the outbreak isolates. Results obtained using the outbreak-specific PCR were confirmed by WGS.
Environment screening
In total, 10 of 57 environmental samples were tested positive for ESBL-KP. To assess the genetic relatedness between environmental and patient isolates, three environmental isolates (KP-E1, KP-E2, and KP-E3) were selected for WGS (Table 1). The phylogenetic analysis showed that all three isolates clustered with the patient isolates (Fig. 2). Isolate KP-E2 (rehabilitation centre; 2012) was indistinguishable from those of five patients (KP-1, KP-2, KP-4, KP-6 and KP-7), and KP-E1 (rehabilitation centre; 2012) and KP-E3 (university hospital; 2013) differed by only up to 3 SNPs from all other patient isolates (see Supplementary Table S1 online).
Resistome
Coding sequences associated with resistance were identified to elucidate the drug-resistance mechanisms of outbreak-associated isolates. Table 2 shows those genes detected in the outbreak isolates. All outbreak isolates shared the same resistome. The ESBL gene blaCTX-M-15 and the non-ESBL gene blaSHV-28 were detected on the plasmid and chromosome, respectively. The detection of acquired drug-resistance genes allowed us to explain the phenotypical resistance profile. Further analyses identified non-synonymous SNPs in acrR (A165T) and ramR (G56A), resulting in an amino-acid substitution in AcrR (K55N) and RamR (A19V). These proteins are regulators of AcrAB-TolC and mutations in these regulators may cause overexpression of AcrAB resulting in MDR phenotypes23. Analysis of three well-described mutational hotspots associated with resistance to quinolone/fluoroquinolone showed non-synonymous nucleotide changes in gyrA and parC genes, resulting in amino-acid substitutions in DNA gyrase (S83F, D87A) and topoisomerase IV (S80I)24.
Virulence factors
All outbreak isolates shared the same virulence factors. Table 3 shows the virulence factors identified in the outbreak clone, which were highly similar with those identified in the ST15 clade II isolate PMK1 from Nepal. Two hypervirulence-related iron uptake systems (ABC transporter Kfu and Yersinia high-pathogenicity island)25, were found in the outbreak clone and PMK1 but not in the non-outbreak isolate KP-33P (clade II). Although both the outbreak clone and PMK1 harboured a genomic island carrying the Yersinia high-pathogenicity island (encoding yersiniabactin) and a type IV(A) secretion system, the island’s compositions and its insertion site were rather different (see Supplementary Fig. S3). The BLASTn result suggests that the genomic island of the outbreak clone may have been acquired from an Escherichia coli stain ED 1a (Table 3). As the other ST15 clade I isolates, the capsular genotype of the outbreak clone was wzi24, associated with serotype K24 (http://bigsdb.web.pasteur.fr/klebsiella/klebsiella.html). This was different from that of clade II isolates PMK1 and KP-33P, which was wzi93, associated with serotype K60 (Fig. 3).

The cps regions of K. pneumoniae PMK1 and KP-33P (non-outbreak isolate of this study) were identical. The cps region of a ST15 strain was retrieved from GenBank. The gradients (dark to pale) of the alignment region represent the percentage of sequence identity between samples defined by BLASTn.
Discussion
Controlling the dissemination of multidrug-resistant pathogens remains one of the major public health challenges in the 21st century. Here, we share our experience in managing a regional outbreak caused by a HiRiC of ESBL-positive K. pneumoniae ST15. This lineage has previously been implicated in hospital outbreaks worldwide4,22,26,27. The regional and temporal dimension of this outbreak was initially unexpected but became visible when WGS data were compared with a locally available database. The outbreak involved patients from three institutions in two cities within an 80-km distance located in the same province of the Netherlands and may well have gone beyond the reach of our regional infection control capacity. The high infection rate of the outbreak clone in patients and the rebound of two patients a year after the start of the outbreak are indicators for the virulence and tenacity of this clone fulfilling the criteria of a HiRiC28,29,30.
The strength of WGS can be fully appreciated when considering that no obvious epidemiological association existed between a sporadic patient in 2013 (patient 11) and patients belonging to a transmission cluster one year before. A compelling degree of sequence homology consisting of 2–4 SNPs difference in the core genome between isolates from unrelated patients, led us to suggest a link, which was subsequently vindicated by epidemiological investigation. The results showed that high-resolution epidemiological typing could support infection control initiatives in unambiguously clustering patients colonized or infected with bacterial species otherwise abundant in clinical specimen.
We demonstrated that tailor-made diagnostic markers by identifying genomic signatures could highly improve the efficiency of infection control, especially for HiRiCs that may be perpetuated in health care networks. Indeed, the outbreak-specific PCR allowed us to rapidly and undoubtedly identify another “sporadic” patient (patient 12) more than one year after the start of the outbreak. Most importantly, this method enables to distinguish outbreak isolates from non-outbreak isolates even if they shared the same sequence type (i.e. ST15 isolate KP-33P). These advantages facilitate the early detection of patients, which can be greatly helpful for the prevention of clonal spread and patient treatments.
The higher resolution of WGS is also an advantage on global-scale epidemiological typing comparing to conventional methods (e.g. MLST). This greatly helps us to dissect the population structure of opportunistic bacterial pathogens by identifying HiRiCs, i.e. clonal lineages of particular public health importance. Recent work based on WGS analysis reveals that K. pneumoniae ST258, another HiRiC associated with the global dissemination KPC carbapenemase, is actually composed of two distinct genetic clades, which challenges conventional hypotheses on clonal descent31. Similarly, our study shows that the population structure of ST15 may be diverse, as two clades were formed by the ST15 isolates analysed. Full understanding of the ST15 genomic population structure will be helpful for the infection management of this HiRiC. Notably, the non-outbreak ST15 isolate KP-33P obtained in 2010 in the UMCG clustered with a ST15 NDM-1-producing Nepali outbreak isolate PMK1 (82 SNPs), suggesting that they share a common ancestor.
Noteworthy, ST15 clades I and II carried two different capsular serotypes (K24 and K60), respectively. This is similar as the recent finding that ST258 clades I and II were mainly differentiated by an ~215-kb region including cps genes31. Capsular serotypes are known to be associated with different virulence-associated traits (e.g. immune evasion and biofilm formation) in carbapenem-resistant K. pneumoniae32. The role of the different capsular serotypes in the pathogenicity of ST15 should be studied in the future to allow improved risk-assessment (i.e. identify hypervirulence- and/or epidemic-related capsule serotypes) and novel therapy (i.e. use of anti-capsule antibodies). Additionally, a Yersinia high-pathogenicity island encoding yersiniabactin was identified in the outbreak clone. This factor was also present in the Nepali outbreak isolate PMK1, but not in the non-outbreak ST15 isolate KP-33P. A recent study showed that yersiniabactin is essential for K. pneumoniae to become an effective respiratory pathogen33. This may be one of reasons for the high infection rate (~58.3%) observed during the outbreak and further suggests that the capacity to rapidly acquire virulence factors is important to become a HiRiC.
Analysis of the resistome identified a set of plasmid-borne resistance genes (blaCTX-M-15-blaTEM-1-blaOXA-1-aac(6′)-Ib-cr-qnrB1) in the outbreak clone that was found in Enterbacteriaceae populations spreading throughout Europe34,35,36,37,38. Acquisition of such resistome may confer an advantage to HiRiCs, thereby stimulating their emergence. Indeed, a highly similar resistome was identified in a new K. pneumoniae clone (ST1427) causing a nosocomial outbreak in our hospital39. Therefore, active surveillance of epidemic plasmid with certain resistome will be helpful in preventing HiRiCs dissemination.
Our study can be useful for making appropriate intervention/prevention strategies for HiRiCs in the future. First of all, the capacity to contaminate the environment facilitates the dissemination of the outbreak clone. Therefore, complete decontaminations, including cleaning and disinfection of surfaces likely to be contaminated by patients (e.g. bed and toilets) are extremely important for outbreak prevention and control. Secondly, frequent screening of patients, especially those receiving long-term healthcare and/or extensive antibiotic treatment, is required for controlling and preventing transmissions. Furthermore, extensive collaboration on diagnostics and infection prevention within a healthcare network40, is important for efficient infection prevention and control. For this, a shared database containing information on characteristics of circulating clones and data of intra- as well as inter-hospital patient transfer would be of great help.
In summary, our study highlights challenges in managing the outbreak of K. pneumoniae HiRiCs, suggesting the necessity of frequent surveillance and inter-institutional collaborations for infection/outbreak prevention and control.
Additional Information
How to cite this article: Zhou, K. et al. Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci. Rep. 6, 20840; doi: 10.1038/srep20840 (2016).
Change history
24 March 2016
The HTML version of this paper was updated after publication, following a technical error that resulted in the links to Supplementary Figures 1-3 being omitted. This has now been corrected; the PDF version of the paper was correct from the time of publication.
References
Boucher, H. W. et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12, doi: 10.1086/595011 (2009).
Holt, K. E. et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 112, E3574–3581, doi: 10.1073/pnas.1501049112 (2015).
Breurec, S. et al. Klebsiella pneumoniae resistant to third-generation cephalosporins in five African and two Vietnamese major towns: multiclonal population structure with two major international clonal groups, CG15 and CG258. Clin. Microbiol. Infect. 19, 349–355, doi: 10.1111/j.1469-0691.2012.03805.x (2013).
Damjanova, I. et al. Expansion and countrywide dissemination of ST11, ST15 and ST147 ciprofloxacin-resistant CTX-M-15-type β-lactamase-producing Klebsiella pneumoniae epidemic clones in Hungary in 2005— the new ‘MRSAs’? J. Antimicrob. Chemother. 62, 978–985, doi: 10.1093/jac/dkn287 (2008).
Hrabák, J. et al. International clones of Klebsiella pneumoniae and Escherichia coli with extended-spectrum beta-lactamases in a Czech hospital. J. Clin. Microbiol. 47, 3353–3357, doi: 10.1128/JCM.00901-09 (2009).
Lee, M. Y. et al. High prevalence of CTX-M-15-producing Klebsiella pneumoniae isolates in Asian countries: diverse clones and clonal dissemination. Int. J. Antimicrob. Agents. 38, 160–163, doi: 10.1016/j.ijantimicag.2011.03.020 (2011).
Rodrigues, C. et al. Expansion of ESBL-producing Klebsiella pneumoniae in hospitalized patients: a successful story of international clones (ST15, ST147, ST336) and epidemic plasmids (IncR, IncFIIK). Int. J. Med. Microbiol. 304, 1100–1108, doi: 10.1016/j.ijmm.2014.08.003 (2014).
Munoz-Price, L. S. et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 13, 785–796, doi: 10.1016/S1473-3099(13)70190-7 (2013).
Snitkin, E. S. et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 4, 148ra116, doi: 10.1126/scitranslmed.3004129 (2012).
Willemsen, I. et al. Trends in Extended Spectrum Beta-Lactamase (ESBL) Producing Enterobacteriaceae and ESBL Genes in a Dutch Teaching Hospital, Measured in 5 Yearly Point Prevalence Surveys (2010–2014). PLoS One. 10, e0141765, doi: 10.1371/journal.pone.0141765 (2015).
Zhou, K. et al. The mosaic genome structure and phylogeny of Shiga toxin-producing Escherichia coli O104:H4 is driven by short-term adaptation. Clin. Microbiol. Infect. 21, 468.e7–468.e18, doi: 10.1016/j.cmi.2014.12.009 (2015).
Boetzer, M. et al. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579, doi: 10.1093/bioinformatics/btq683 (2011).
Boetzer, M. & Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 13, R56, doi: 10.1186/gb-2012-13-6-r56 (2012).
Aziz, R. K. et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics 9, 75, doi: 10.1186/1471-2164-9-75 (2008).
Li, B., Zhao, Y., Liu, C., Chen, Z. & Zhou, D. Molecular pathogenesis of Klebsiella pneumoniae . Future Microbiol. 9, 1071–1081, doi: 10.2217/fmb.14.48 (2014).
Ramos, P. I. et al. Comparative analysis of the complete genome of KPC-2-producing Klebsiella pneumoniae Kp13 reveals remarkable genome plasticity and a wide repertoire of virulence and resistance mechanisms. BMC Genomics. 15, 54, doi: 10.1186/1471-2164-15-54 (2014).
Brisse, S. et al. wzi Gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J. Clin. Microbiol. 51, 4073–4078, doi: 10.1128/JCM.01924-13 (2013).
Assefa, S. et al. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 25, 1968–1969, doi: 10.1093/bioinformatics/btp347 (2009).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690, doi: 10.1093/bioinformatics/btl446 (2006).
Darling, A. E., Mau, B. & Perna, N.T. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147, doi: 10.1371/journal.pone.0011147 (2010).
Shen, Z. et al. MPprimer: a program for reliable multiplex PCR primer design. BMC Bioinformatics. 11, 143, doi: 10.1186/1471-2105-11-143 (2010).
Stoesser, N. et al. Genome sequencing of an extended series of NDM-producing Klebsiella pneumoniae isolates from neonatal infections in a Nepali hospital characterizes the extent of community- versus hospital-associated transmission in an endemic setting. Antimicrob. Agents Chemother. 58, 7347–7357, doi: 10.1128/AAC.03900-14 (2014).
Bialek-Davenet, S. et al. In vitro selection of ramR and soxR mutants overexpressing efflux systems by fluoroquinolones as well as cefoxitin in Klebsiella pneumoniae . Antimicrob. Agents Chemother. 55, 2795–2802, doi: 10.1128/AAC.00156-11 (2011).
Deguchi, T. et al. Alterations in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in quinolone-resistant clinical isolates of Klebsiella pneumoniae . Antimicrob. Agents Chemother. 41, 699–701, (1997).
Shon, A. S., Bajwa, R. P. & Russo, T. A. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 4, 107–118, doi: 10.4161/viru.22718 (2013).
Chung, The, H. et al. A high-resolution genomic analysis of multidrug-resistant hospital outbreaks of Klebsiella pneumoniae. EMBO Mol. Med. 7, 227–239, doi: 10.15252/emmm.201404767 (2015).
Nielsen, J. B. et al. Identification of CTX-M15-, SHV-28-producing Klebsiella pneumoniae ST15 as an epidemic clone in the Copenhagen area using a semi-automated REP-PCR typing assay. Eur. J. Clin. Microbiol. Infect. Dis. 30, 773–778, doi: 10.1007/s10096-011-1153-x (2011).
Baquero, F. & Coque, T. M. Multilevel population genetics in antibiotic resistance. FEMS Microbiol. Rev. 35, 705–706, doi: 10.1111/j.1574-6976.2011.00293.x (2011).
Willems, R. J. et al. Population biology of Gram-positive pathogens: high-risk clones for dissemination of antibiotic resistance. FEMS Microbiol. Rev. 35, 872–900, doi: 10.1111/j.1574-6976.2011.00284.x (2011).
Woodford, N., Turton, J. F. & Livermore, D. M. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol. Rev. 35, 736–755, doi: 10.1111/j.1574-6976.2011.00268.x (2011).
Deleo, F. R. et al. Molecular dissection of the evolution of carbapenem-resistant multilocus sequence type 258 Klebsiella pneumoniae . Proc. Natl. Acad. Sci. USA 111, 4988–4993, doi: 10.1073/pnas.1321364111 (2014).
Diago-Navarro, E. et al. Carbapenem-resistant Klebsiella pneumoniae exhibit variability in capsular polysaccharide and capsule associated virulence traits. J. Infect. Dis. 210, 803–813, doi: 10.1093/infdis/jiu157 (2014).
Bachman, M. A. et al. Klebsiella pneumoniae yersiniabactin promotes respiratory tract infection through evasion of lipocalin 2. Infect. Immun. 79, 3309–3316, doi: 10.1128/IAI.05114-11 (2010).
Dolejska, M. et al. Plasmid content of a clinically relevant Klebsiella pneumoniae clone from the Czech Republic producing CTX-M-15 and QnrB1. Antimicrob. Agents Chemother. 57, 1073–1076, doi: 10.1128/AAC.01886-12 (2013).
Filippa, N. et al. Outbreak of multidrug-resistant Klebsiella pneumoniae carrying qnrB1 and blaCTX-M15 in a French intensive care unit. Ann. Intensive Care 3, 18, doi: 10.1186/2110-5820-3-18 (2013).
Huang, T. W. et al. Copy number change of the NDM-1 sequence in a multidrug-resistant Klebsiella pneumoniae clinical isolate. PLoS ONE. 8, e62774. doi: 10.1371/journal.pone.0062774 (2013).
Bialek-Davenet, S. et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 20 1812–1820, doi: 10.3201/eid2011.140206 (2014).
Machado, E. et al. Dissemination in Portugal of CTX-M-15-, OXA-1-, and TEM-1-producing Enterobacteriaceae strains containing the aac(6′)-Ib-cr gene, which encodes an aminoglycoside- and fluoroquinolone-modifying enzyme. Antimicrob. Agents Chemother. 50, 3220–3221, doi: 10.1128/AAC.00473-06 (2006).
Zhou, K. et al. Characterization of a CTX-M-15 producing Klebsiella pneumoniae outbreak strain assigned to a novel sequence type (1427). Front. Microbiol. 6, 1250, doi: 10.3389/fmicb.2015.01250 (2015).
Donker, T., Wallinga, J. & Grundmann, H. Dispersal of antibiotic-resistant high-risk clones by hospital networks: changing the patient direction can make all the difference. J. Hosp. Infect. 86, 34–41, doi: 10.1016/j.jhin.2013.06.021 (2014).
Acknowledgements
We thank Mariano Ciccolini for generating the figure of the intra-hospital patient transfer. This work was done in collaboration with the ESCMID Study Group on Molecular Diagnostics (ESGMD) and the ESCMID Study Group on Molecular Epidemiological Markers (ESGEM), Basel, Switzerland. This study was partly supported by the Interreg IVa-funded projects EurSafety Heath-net (III-1-02 = 73) and by a University Medical Center Groningen Healthy Ageing Pilots grant.
Author information
Authors and Affiliations
Contributions
K.Z., M.L., R.H.D., J.P.A., E.G.C.R., J.L.T.F., H.G., J.W.A.R. and A.W.F. participated in the design and/or discussion of the study. K.Z., M.L. and E.G.C.R. carried out the major experiments. K.Z., M.L. and R.H.D. analyzed the data. K.Z., M.L. and J.W.A.R. wrote the paper. M.T., J.L.T.F., H.G., J.W.A.R. and A.W.F. revised it for important intellectual improvement. All authors read and approved the final version to be published
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhou, K., Lokate, M., Deurenberg, R. et al. Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae. Sci Rep 6, 20840 (2016). https://doi.org/10.1038/srep20840
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20840
This article is cited by
-
Contact investigations for antibiotic-resistant bacteria: a mixed-methods study of patients’ comprehension of and compliance with self-sampling requests post-discharge
Antimicrobial Resistance & Infection Control (2023)
-
A novel and improved selective media for the isolation and enumeration of Klebsiella species
Applied Microbiology and Biotechnology (2022)
-
The diagnostic value of metagenomic next⁃generation sequencing in infectious diseases
BMC Infectious Diseases (2021)
-
Shotgun-metagenomics based prediction of antibiotic resistance and virulence determinants in Staphylococcus aureus from periprosthetic tissue on blood culture bottles
Scientific Reports (2021)
-
Enterococcus faecium: from microbiological insights to practical recommendations for infection control and diagnostics
Antimicrobial Resistance & Infection Control (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
