
Abstract
The endoplasmic reticulum (ER) has a sophisticated protein quality control system for the efficient folding of newly synthesized proteins. In this system, a variety of N-linked oligosaccharides displayed on proteins serve as signals recognized by series of intracellular lectins. Glucosidase II catalyzes two-step hydrolysis at α1,3-linked glucose–glucose and glucose–mannose residues of high-mannose-type glycans to generate a quality control protein tag that is transiently expressed on glycoproteins and recognized by ER chaperones. Here we determined the crystal structures of the catalytic α subunit of glucosidase II (GIIα) complexed with two different glucosyl ligands containing the scissile bonds of first- and second-step reactions. Our structural data revealed that the nonreducing terminal disaccharide moieties of the two kinds of substrates can be accommodated in a gourd-shaped bilocular pocket, thereby providing a structural basis for substrate-binding specificity in the two-step deglucosylation catalyzed by this enzyme.
Similar content being viewed by others
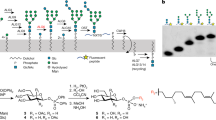

Introduction
Glycoproteins in the early secretory pathway are subject to quality control, in which their N-linked glycans play key roles as protein maturation and quality control tags1,2,3,4,5,6,7. In the endoplasmic reticulum (ER), the triantennary tetradecasaccharide Glc3Man9GlcNAc2 is attached to nascent proteins as a common precursor of N-glycans. This high-mannose-type oligosaccharide has three nonreducing terminal branches (termed D1, D2 and D3, Fig. 1a) and embeds various carbohydrate epitopes (glycotopes) recognized by disparate lectins operating as molecular chaperones, cargo receptors and degradation mediators. These glycoprotein fate determinants are sequentially exposed by the actions of series of glucosidases and mannosidases. The D1 branch of the initial glycoform is capped by a triglucosyl moiety, Glc-α1,2-Glc-α1,3-Glc. Glucosidase I removes the outermost α1,2-linked glucose from the D1 branch8,9 and then glucosidase II (GII) trims second and third α1,3-linked glucose residues by catalyzing hydrolyses at the Glc-α1,3-Glc and Glc-α1,3-Man glycosidic linkages6,8,10,11. The monoglucosylated glycoform transiently expressed during the two-step deglucosylation by GII serves as a signal recognized by ER lectins, calnexin (CNX) and calreticulin (CRT)12,13,14,15 which form complexes with a folding catalyst, protein disulfide isomerase family protein ERp5716,17. Complete deglucosylation by GII prompts the disengagement of glycoproteins from chaperone complexes for anterograde transport to the Golgi apparatus if they are successfully folded18,19,20,21. In contrast, glycoproteins yet to be folded are reglucosylated by the folding sensor enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT), giving rise to their monoglucosylated glycoform and thereby enabling them to revisit the chaperone complex22,23,24. Thus, N-glycans act as a timer in the determination of glycoprotein fates.

Overall structure of the glucosidase II α subunit.
(a) Schematic representation of Glc3Man9GlcNAc2 showing the nomenclature of oligosaccharide residues and branches. Glucose residues trimmed by GII are shown in red. (b) Ribbon model of GIIα is represented with positions of N and C termini and individual domains. The individual domains are colored as the following: N-terminal domain (yellow), subdomain B1 (hot pink), β8α8 barrel domain (red), subdomain B2 (purple), subdomain B3 (orange), proximal C-terminal domain (blue) and distal C-terminal domain (green). The characteristic N-terminal segment is colored in cyan. The catalytic residues are shown as pale cyan sphere models.
GII consists of a 110-kDa catalytic α subunit (GIIα) belonging to the glycosyl hydrolase 31 family (GH31, EC. 3.2.1.84) and a 60-kDa noncatalytic regulatory β subunit (GIIβ) having a flexible extended structure that contains a mannose 6-phosphate receptor homology (MRH) domain25,26,27,28. Among GH31 enzymes, only GII shows α1,3-glucosidase activity. Several crystal structures of GH31 enzymes have been reported29,30,31,32,33,34, including maltase–glucoamylase and sucrase–isomaltase (EC. 3.2.1.20), which are specific for α1,4- and α1,4-/1,6-linked glucosyl substrates, respectively. However, no structural information has so far been available for GII except for the recently reported NMR and crystal structures of its MRH domain35,36, which have provided structural insights into its binding specificity to the D3 branch37,38,39. Thus, the structural basis for the two-step glucose trimming reactions catalyzed by GII remains unclear. Here we performed an X-ray crystallographic study of GIIα to elucidate its substrate recognition mechanism.
Results and Discussion
Overall structure of the catalytic α subunit of glucosidase II
Considering the potential protein stability, Chaetomium thermophilum, a thermophilic fungus, which survives at temperatures of up to 60 °C40, was selected as organism source for the structural study of GIIα. It has been demonstrated that the closely-related species such as a fission yeast Schizosaccharomyces pombe and a filamentous fungus Aspergillus oryzae possess an enzymatically active glucosidase II with the same substrate specificity as that of the mammalian counterparts35,36,37,39. We expressed the recombinant GIIα (residues 31–977) in Escherichia coli and crystallized it by the hanging-drop vapor diffusion method. The crystal belongs to space group R32 with one molecule per asymmetric unit. The final model of GIIα refined to 1.40 Å resolution has an Rwork of 15.4% and Rfree of 17.5% (Table 1).
The overall structure of GIIα is composed of four major domains and three subdomains: N-terminal domain (residues 31–384), subdomain B1 (residues 207–256), β8α8 barrel domain (residues 385–737), subdomain B2 (residues 484–526), subdomain B3 (residues 559–581), proximal C-terminal domain (residues 738–823) and distal C-terminal domain (residues 824–977) (Fig. 1b and Supplemental Fig. S1). This fold is essentially identical to that of other GH31 α-glucosidases29,30,31,32,33,34 (Supplemental Fig. S2).
The N-terminal domain has a β sandwich of four anti-parallel β sheets and is composed of 17 β strands. This domain contains a characteristic 14-residue-long α helix (termed α1) at the N-terminus and two short α helices at the β4–5 loop as compared with the other GH31 enzymes29,30,31,32,33,34. The N-terminal segment is highly diverse among GH31 α-glucosidases (Supplemental Fig. S2). The α1 helix covers a β sheet comprising β12, β17 and β18, and, the preceding 11-residue-long segment is accommodated in the putative active site pocket of β8α8 barrel domain, suggesting its involvement in substrate binding. The cysteine residues (Cys39–Cys45) in the N-terminal segment form a disulfide bond. In contrast, the N-terminal segment in the other GH31s is situated outward with respect to their putative active site pocket. Furthermore, a β14–15 long loop, the so-called “N-loop” (Supplemental Fig. S1), forms part of the putative active site pocket as in the other GH31s29,30,31,32,33,34. A unique subdomain (termed B1) is found in the N-terminal domain of GIIα but not in the other GH31s (Supplemental Fig. S2). This subdomain, containing a short β-hairpin (β10–β11), is inserted into the β9–12 loop and is in contact mainly with the N-terminal segment and subdomain B3. In subdomain B1, residues 215–235 are completely disordered, suggesting its flexible nature. Among the known GH31 enzymes, only GII forms an α/β hetero-dimeric structure. It is thus plausible that this unique subdomain is involved in the interaction with the β subunit.
The 352-residue-long β8α8 barrel constitutes the major domain of GIIα (approximately 40%) and forms the putative active-site pocket together with the N-loop. The tris(hydroxymethyl)aminomethane (Tris) molecule derived from the crystallization reagent, also known as an α-glucosidase inhibitor41, was accommodated in the putative active-site pocket located at the center of β8α8 barrel domain. The details of the interaction mode will be explained in the next section. The β8α8 barrel domain has two inserted subdomains, B2 and B3, which are also parts of the active-site pocket. The subdomain B2, containing one β sheet (β23-β24) and one α helix (α7), is inserted into the β23–α8 loop, whereas the subdomain B3, having no typical structure element, is inserted into the β25–α9 loop.
The proximal C-terminal domain is composed of three six-stranded anti-parallel β sheets. In contrast, the distal C-terminal domain consists of two ten-stranded anti-parallel β sheets and three small α helices. In this domain, a continuous twisted β strand (β43) connects the β sheets with β42 and β44, forming a β-barrel-like structure. These C-terminal domains appear to be responsible for the stabilization of the β8α8 barrel domain rather than for substrate binding, given that no interactions between these C-terminal domains and the active-site pocket were observed.
Active site of GIIα
To identify the active site of GII, we determined the crystal structure of GIIα with its inhibitor, 1-deoxynojirimycin (DNJ). The final model of DNJ-bound GIIα refined to 1.60 Å resolution has an Rwork of 15.5% and Rfree of 18.3% (Table 1). As expected, the DNJ molecule was accommodated in the putative active-site pocket including a WiDMNE consensus motif of the GH31 subgroup 1 (the i position is variable and occupied by an asparagine residue in GIIα) in the β8α8 barrel domain32 (Fig. 1b and Supplemental Fig. S1). The DNJ molecule adopts a 4C1 chair conformation in the crystal. All hydroxyl groups and the amide group of DNJ were extensively involved in binding through hydrogen bonds with Asp443, Asp556, Arg617, Asp633 and His691 (Supplemental Fig. S3a). In addition, Asp482 and Asp662 formed water-mediated hydrogen bonds. The Tris molecule was bound to the pocket in the 1.40-Å-resolution crystal structure. Similar to DNJ, it interacted with Asp443, Asp556, Arg617, Asp633 and His691 through hydrogen bonds (Supplemental Fig. S3b). The residues involved in their interactions are highly conserved in GH31 α-glucosidases and the inhibitor-binding modes of GIIα are almost identical to those of other GH31 α-glucosidases29,30,31,32,33,34. On the basis of these results together with previous biochemical data28,42, we concluded that GII shares a common catalytic mechanism with other GH31 α-glucosidases through the conserved active site. GH31-family enzymes are known to be retaining α-glycosidases, which hydrolyze glycoside bonds with the retention of anomeric configuration by an acid/base-catalyzed mechanism involving a covalent glycosyl-enzyme intermediate34,43. It is supposed that the Asp556 and Asp633 act as a catalytic nucleophile and acid/base, respectively. We also confirmed that the active-site residues of the thermophilic fungus GII are highly conserved, indicating a common enzymatic mechanism across species (Supplemental Fig. S1).
Substrate recognition of GIIα
To elucidate the two-step enzymatic reaction mechanisms of GII, we performed the structural determination of substrate-bound complexes of GIIα using its inactive mutant in which the catalytic residue Asp556 was substituted with alanine. We used Glc-α1,3-Glc (α3-Glc2) and Glc-α1,3-Man-α1,2-Man (Glc1Man2) corresponding to Glc(G2)-Glc(G3) and Glc(G3)-Man(D1)-Man(C) in Glc3Man9GlcNAc2, respectively (Fig. 1a). The final model of the α3-Glc2-bound GIIα refined to 2.40 Å resolution has an Rwork of 14.8% and Rfree of 19.9%, whereas that of the Glc1Man2-bound form refined to 2.30 Å resolution has an Rwork of 14.4% and Rfree of 18.9% (Table 1). In substrate-bound forms, two residues (Val31 and Phe32) at the N terminus are disordered, suggesting the flexible nature of the N-terminal segment near the active site.
The α3-Glc2 ligand containing the scissile bond of the first reaction of GII was accommodated in a gourd-shaped bilocular pocket (Fig. 2a). Such a bilocular active-site pocket is also found in other GH31 enzymes, i.e., N-terminal domains of maltase–glucoamylase30 and sucrase-isomaltase29 with short-chain substrate specificities30,44. Two glucose residues were clearly visible in the electron density map and were traced as Glc-α1,3-Glc (Fig. 2b). Although anomeric stereochemistry of the reducing-end sugar residue is usually in the equilibrium of the α and β configurations that are visualized in high-resolution crystal structures as exemplified by an 1.80-Å crystal structure of GH95 1,2-α-L-fucosidase45, the alternative anomeric configurations of the glucose residue was not clearly observed in the electron density map in the Glc-α1,3-Glc-bound GIIα crystal structure (Fig. 2b) probably due to its medium resolution (2.4 Å). In the present crystal structure, α configuration that was more clearly identified in the electron density map was modeled. The position of the nonreducing end Glc(G2) residue is almost identical to that of DNJ (Supplemental Fig. S3a). The Glc(G2) also adopted a chair 4C1 conformation as in DNJ. The disaccharide ligand binds to the pocket with the nonreducing terminal Glc(G2) residue interacting with the deeply buried −1 subsite and the reducing Glc(G3) occupying the surface-proximal +1 subsite (Fig. 2a), explaining its exo-α1,3-glucosidase activity6,8,10,11. In the −1 subsite, all the hydroxyl groups of the Glc(G2) residue interacted with Asp443, Arg617, Asp633 and His691. Additionally, Asp482, Trp517, Trp630 and Asp662 formed hydrogen bonds via water molecules and Trp415 formed a hydrophobic interaction. Among these residues, only Trp517 is located in the subdomain B3, whereas others are located in the β8α8 barrel domain. In the +1 subsite, the 4-OH and 6-OH groups of the glucose residue form hydrogen bonds with Asp303 (N-loop) and Arg617. Remarkably, the substrate-binding residues Asp303 (N-loop), Trp517 (subdomain B3) and Arg617 (β8α8 barrel domain) interacted with Ser561, Phe563 and Glu559, respectively, in the subdomain B2, suggesting their contribution to the stabilization of the substrate-binding pocket.

Substrate-binding site of GIIα.
Substrate-binding pocket represented by sliced surface models and detailed substrate–interaction network with potential hydrogen bonds of GIIα are indicated: (a,b) Glc-α1,3-Glc-bound form, (c,d) Glc-α1,3-Man-bound form. Omit Fo-Fc electron density map of Glc-α1,3-Glc (b), Glc-α1,3-Man (d) and bound water molecules contoured at 2.0 σ. Bound sugar ligands and residues involved in ligand binding are shown in stick models. Water molecules are shown in sphere models. Dashed lines indicate potential hydrogen bonds.
The disaccharide part of the Glc1Man2 ligand was accommodated in the active-site pocket in almost the same manner as α3-Glc2 (Fig. 2c). The Glc(G3) and Man(D1) residues were clearly visible in the electron density map and were assigned as Glc-α1,3-Man with a 4C1 chair conformation, corresponding to the scissile site of the second reaction catalyzed by GII, whereas the reducing-terminal Man(C) residue was almost completely disordered (Fig. 2d). This observation is explained as follows: Man(C) is turned toward the solvent, confirming that the substrate-binding pocket of GIIα is composed of only two subsites, −1 and +1, as in bilocular pockets of the N-terminal domains of maltase–glucoamylase30 and sucrase-isomaltase29. A difference between the two substrate-bound GIIα structures was observed with respect to the direction of the 2-OH group of the carbohydrate residue accommodated in the +1 subsite. In the Glc(G2)-Glc(G3)-bound complex, the equatorial 2-OH group of Glc(G3) is not involved in interaction (Fig. 2a,b), whereas the axial 2-OH group of Man(D1) forms a hydrogen bond with Asp633 in the Glc(G3)-Man(D1)-bound complex (Fig. 2c,d), suggesting its contribution to enhanced affinity. Previous kinetics data demonstrated that first cleavage (Glc2Man9GlcNAc2 → Glc1Man9GlcNAc2) catalyzed by GII αβ heterodimer is significantly faster than second cleavage (Glc1Man9GlcNAc2 → Man9GlcNAc2) in vitro10. Based on the results together with our structural data, we speculate that the delayed second cleavage reaction is attributed to the enhanced interaction through 2OH group of Man(D1) in the +1 subsite. Taken together, our structural data revealed that GIIα can recognize two kinds of glucosylated substrates, Glc-α1,3-Glc and Glc-α1,3-Man, via a bilocular substrate-binding pocket with a tolerant +1 subsite.
Our crystal structures also suggest that the two-step glucose trimming reactions catalyzed in the GII active-site pocket do not successively proceed by virtue of its gourd-shaped architecture. The first glucose product must be eliminated from the deeply buried −1 subsite through the +1 subsite prior to the accommodation of the second cleavage site of the substrate. It is plausible that the delayed second cleavage reaction has a functional advantage, offering glycoproteins a time window for chaperone-mediated folding, given that the presence of a monoglucose residue on high-mannose-type oligosaccharides is essential for interaction with the folding machinery6,13.
In summary, our crystallographic data provide the first structural insights into glycoprotein processing via catalysis by GII of two-step glucose trimming reactions involved in the ER quality control system. The present example is also the first of the structural determination of a GH31 enzyme showing α1,3-glucosidase activity.
Materials and Methods
Protein expression and purification
Total RNA extraction and cDNA synthesis from Chaetomium thermophilum var. thermophilum La Touche (DSMZ 1495) were performed as previously described46. Full-length GIIα cDNA was cloned by PCR using sequence data derived from a C. thermophilum genome40. The GIIα inactive mutant D556A in which the catalytic residue Glu556 is mutated to alanine was also constructed. Recombinant wild-type and D556A GIIα proteins were produced as glutathione S-transferase (GST)-fused forms. The full-length GIIα (residues 31–977), excluding the signal peptide, was amplified by PCR and subcloned into the EcoRI and XbaI sites of a modified pCold-GST vector46. Recombinant proteins were produced in E. coli BL21-CodonPlus (DE3, Agilent Technologies) according to the standard protocols provided by the manufacturer (Takara Bio Inc.). The selenomethionine (SeMet)-labeled GIIα was expressed in E. coli B834 (DE3) using M9 minimal medium supplemented with SeMet instead of Met. GST-fused proteins were captured on glutathione-SepharoseTM columns (GE Healthcare) and extensively washed with 20 and 10 column volumes of 20 mM Tris-HCl (pH 7.5) containing 600 and 150 mM NaCl, respectively. GIIα proteins were then eluted from the columns by addition of tobacco etch virus (TEV) protease with 12 h incubation at 4 °C. The resultant nontagged GIIα proteins were further purified by size-exclusion chromatography (Superdex-200, GE Healthcare).
Crystallization, X-ray data collection and structure determination
The GIIα protein (8 mg/mL) was dissolved in 20 mM Tris-HCl (pH 7.5) and 150 mM NaCl and the native crystals were obtained in a buffer containing 1.7 M sodium malonate and 100 mM Tris-HCl (pH 7.5) by incubation at 20 °C for 1 week. The SeMet-substituted crystals were grown in a buffer containing 1.8 M ammonium citrate tribasic and 0.1 M Tris-HCl (pH 7.0). The 1-deoxynojirimycin (DNJ, Sigma-Aldrich)-bound crystals were prepared by soaking in a crystallization buffer containing 1.2 M sodium citrate tribasic, 0.1 M Tris-HCl (pH 7.0) and 2.5 mM DNJ for 30 min. To obtain substrate-bound complexes, the inactive mutant D556A-GIIα crystals were obtained by the equilibration of a protein solution with 1.2 M sodium citrate tribasic and 0.1 M Tris-HCl (pH 8.0). The mutant crystals were soaked with into a crystallization buffer containing 5 mM Glc-α1,3-Man-α1,2-Man (Glc1Man2) or α3-Glc2 (Sigma-Aldrich) for 1 h. α-D-glucopyranosyl-(1 → 3)-α-D-mannopyranosyl-(1 → 2)-α-D-mannopyranose (Glc1Man2) was chemically synthesized as shown in Supplemental Fig. S4. The native crystal was transferred into the reservoir solution and flash-cooled in liquid nitrogen, whereas other crystals were cryoprotected with a soaking buffer supplemented with 20% glycerol. The crystals of GIIα belonged to space group R32 with one molecule per asymmetric unit and diffracted up to a resolution of 1.40 Å (Tris-bound), 1.60 Å (DNJ-bound), 2.30 Å (Glc1Man2-bound) and 2.40 Å (α3-Glc2-bound). Diffraction data were processed with HKL200047. The crystal parameters are shown in Table 1.
The 2.40-Å crystal structure of SeMet-substituted GIIα was solved by the single-wavelength anomalous dispersion (SAD) method using the program Autosol in the Phenix suite48. Using the 1.40-Å native data set, further automated model building and manual model fitting to the electron density maps were performed with ARP/warp49 and COOT50, respectively. The refinement procedure was performed with REFMAC551. The stereochemical quality of the final model was validated with PROCHECK52. The refinement statistics are summarized in Table 1. The molecular graphics were prepared using PyMOL (http://www.pymol.org/).
Additional Information
Accession codes: The coordinates and structural factors of the crystal structures of GIIα have been deposited in the Protein Data Bank under accession numbers 5DKX (Tris-bound), 5DKY (DNJ-bound), 5DKZ (Glc2-bound) and 5DL0 (Glc1Man2-bound).
How to cite this article: Satoh, T. et al. Structural basis for two-step glucose trimming by glucosidase II involved in ER glycoprotein quality control. Sci. Rep. 6, 20575; doi: 10.1038/srep20575 (2016).
References
Ellgaard, L. & Helenius, A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4, 181–91 (2003).
Kato, K. & Kamiya, Y. Structural views of glycoprotein-fate determination in cells. Glycobiology 17, 1031–44 (2007).
Takeda, Y., Totani, K., Matsuo, I. & Ito, Y. Chemical approaches toward understanding glycan-mediated protein quality control. Curr Opin Chem Biol 13, 582–91 (2009).
Lederkremer, G. Z. Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol 19, 515–23 (2009).
Aebi, M., Bernasconi, R., Clerc, S. & Molinari, M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci 35, 74–82 (2010).
D’Alessio, C., Caramelo, J. J. & Parodi, A. J. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin Cell Dev Biol 21, 491–9 (2010).
Kamiya, Y., Satoh, T. & Kato, K. Molecular and structural basis for N-glycan-dependent determination of glycoprotein fates in cells. Biochim Biophys Acta 1820, 1327–37 (2012).
Grinna, L. S. & Robbins, P. W. Substrate specificities of rat liver microsomal glucosidases which process glycoproteins. J Biol Chem 255, 2255–8 (1980).
Deprez, P., Gautschi, M. & Helenius, A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell 19, 183–95 (2005).
Totani, K., Ihara, Y., Matsuo, I. & Ito, Y. Substrate specificity analysis of endoplasmic reticulum glucosidase II using synthetic high mannose-type glycans. J Biol Chem 281, 31502–8 (2006).
Grinna, L. S. & Robbins, P. W. Glycoprotein biosynthesis. Rat liver microsomal glucosidases which process oligosaccharides. J Biol Chem 254, 8814–8 (1979).
Schrag, J. D. et al. The structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol Cell 8, 633–44 (2001).
Caramelo, J. J. & Parodi, A. J. Getting in and out from calnexin/calreticulin cycles. J Biol Chem 283, 10221–5 (2008).
Kozlov, G. et al. Structural basis of carbohydrate recognition by calreticulin. J Biol Chem 285, 38612–20 (2010).
Chouquet, A. et al. X-ray structure of the human calreticulin globular domain reveals a peptide-binding area and suggests a multi-molecular mechanism. PLoS ONE 6, e17886 (2011).
Määttänen, P., Kozlov, G., Gehring, K. & Thomas, D. Y. ERp57 and PDI: multifunctional protein disulfide isomerases with similar domain architectures but differing substrate-partner associations. Biochem Cell Biol 84, 881–9 (2006).
Jessop, C. E., Tavender, T. J., Watkins, R. H., Chambers, J. E. & Bulleid, N. J. Substrate specificity of the oxidoreductase ERp57 is determined primarily by its interaction with calnexin and calreticulin. J Biol Chem 284, 2194–202 (2009).
Hauri, H. P., Kappeler, F., Andersson, H. & Appenzeller, C. ERGIC-53 and traffic in the secretory pathway. J Cell Sci 113 (Pt 4), 587–96 (2000).
Nishio, M. et al. Structural basis for the cooperative interplay between the two causative gene products of combined factor V and factor VIII deficiency. Proc Natl Acad Sci USA 107, 4034–9 (2010).
Wigren, E., Bourhis, J. M., Kursula, I., Guy, J. E. & Lindqvist, Y. Crystal structure of the LMAN1-CRD/MCFD2 transport receptor complex provides insight into combined deficiency of factor V and factor VIII. FEBS Lett 584, 878–82 (2010).
Satoh, T., Suzuki, K., Yamaguchi, T. & Kato, K. Structural basis for disparate sugar-binding specificities in the homologous cargo receptors ERGIC-53 and VIP36. PLoS One 9, e87963 (2014).
Taylor, S. C., Ferguson, A. D., Bergeron, J. J. & Thomas, D. Y. The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol 11, 128–34 (2004).
Caramelo, J. J., Castro, O. A., Alonso, L. G., De Prat-Gay, G. & Parodi, A. J. UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci USA 100, 86–91 (2003).
Totani, K., Ihara, Y., Tsujimoto, T., Matsuo, I. & Ito, Y. The recognition motif of the glycoprotein-folding sensor enzyme UDP-Glc:glycoprotein glucosyltransferase. Biochemistry 48, 2933–40 (2009).
Trombetta, E. S., Fleming, K. G. & Helenius, A. Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry 40, 10717–22 (2001).
Trombetta, E. S., Simons, J. F. & Helenius, A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem 271, 27509–16 (1996).
Munro, S. The MRH domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr Biol 11, R499–501 (2001).
Pelletier, M. F. et al. The heterodimeric structure of glucosidase II is required for its activity, solubility and localization in vivo. Glycobiology 10, 815–27 (2000).
Sim, L. et al. Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J Biol Chem 285, 17763–70 (2010).
Sim, L., Quezada-Calvillo, R., Sterchi, E. E., Nichols, B. L. & Rose, D. R. Human intestinal maltase-glucoamylase: crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J Mol Biol 375, 782–92 (2008).
Ren, L. et al. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2, 827–36 (2011).
Ernst, H. A. et al. Structure of the Sulfolobus solfataricus α-glucosidase: implications for domain conservation and substrate recognition in GH31. J Mol Biol 358, 1106–24 (2006).
Tagami, T. et al. Molecular basis for the recognition of long-chain substrates by plant α-glucosidases. J Biol Chem 288, 19296–303 (2013).
Lovering, A. L., Lee, S. S., Kim, Y. W., Withers, S. G. & Strynadka, N. C. Mechanistic and structural analysis of a family 31 α-glycosidase and its glycosyl-enzyme intermediate. J Biol Chem 280, 2105–15 (2005).
Olson, L. J. et al. Structure of the lectin mannose 6-phosphate receptor homology (MRH) domain of glucosidase II, an enzyme that regulates glycoprotein folding quality control in the endoplasmic reticulum. J Biol Chem 288, 16460–75 (2013).
Olson, L. J. et al. Crystal structure and functional analyses of the lectin domain of glucosidase II: Insights into oligomannose recognition. Biochemistry 54, 4097–111 (2015).
Watanabe, T. et al. Genetic analysis of glucosidase II β-subunit in trimming of high-mannose-type glycans. Glycobiology 19, 834–40 (2009).
Hu, D. et al. Sugar-binding activity of the MRH domain in the ER α-glucosidase II β subunit is important for efficient glucose trimming. Glycobiology 19, 1127–35 (2009).
Stigliano, I. D., Caramelo, J. J., Labriola, C. A., Parodi, A. J. & D’Alessio, C. Glucosidase II β subunit modulates N-glycan trimming in fission yeasts and mammals. Mol Biol Cell 20, 3974–84 (2009).
Amlacher, S. et al. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell 146, 277–89 (2011).
Jorgensen, B. B. & Jorgensen, O. B. Inhibition of barley malt α-glucosidase by Tris(hydroxymethyl)aminomethane and erythritol. Biochim Biophys Acta 146, 167–72 (1967).
Feng, J., Romaniouk, A. V., Samal, S. K. & Vijay, I. K. Processing enzyme glucosidase II: proposed catalytic residues and developmental regulation during the ontogeny of the mouse mammary gland. Glycobiology 14, 909–21 (2004).
Lee, S. S., He, S. & Withers, S. G. Identification of the catalytic nucleophile of the Family 31 α-glucosidase from Aspergillus niger via trapping of a 5-fluoroglycosyl-enzyme intermediate. Biochem J 359, 381–6 (2001).
Quezada-Calvillo, R. et al. Luminal starch substrate “brake” on maltase-glucoamylase activity is located within the glucoamylase subunit. J Nutr 138, 685–92 (2008).
Nagae, M. et al. Structural basis of the catalytic reaction mechanism of novel 1,2-α-L-fucosidase from Bifidobacterium bifidum. J Biol Chem 282, 18497–509 (2007).
Zhu, T., Satoh, T. & Kato, K. Structural insight into substrate recognition by the endoplasmic reticulum folding-sensor enzyme: crystal structure of third thioredoxin-like domain of UDP-glucose:glycoprotein glucosyltransferase. Sci Rep 4, 7322 (2014).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology 276, 307–326 (1997).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–21 (2010).
Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc 3, 1171–9 (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 (2010).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–55 (1997).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993).
Acknowledgements
Diffraction data sets were collected at BL5A and NE3A at the Photon Factory, High Energy Accelerator Research Organization (KEK, Japan) and Osaka University using BL44XU at SPring-8 (Japan). We thank the beamline staff for providing the data collection facilities and support. We also thank Ms. Tong Zhu for her contribution during the early stage of this study. This work was supported in part by the Okazaki ORION project and Grants-in-Aid for Scientific Research (Grant Numbers 24770102, 25121730 to T.S. and 15H02491, 25102008, 24249002 to K.K.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan and by PRESTO project (Grant Number 13417569 to T.S.) from the Japan Science and Technology Agency.
Author information
Authors and Affiliations
Contributions
T.S. and K.K. conceived and designed the experiments; T.S. and T.T. performed the crystallographic experiments; G.Y. and T.Y. performed the chemical synthesis of oligosaccharide ligand; T.S. and K.K. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Satoh, T., Toshimori, T., Yan, G. et al. Structural basis for two-step glucose trimming by glucosidase II involved in ER glycoprotein quality control. Sci Rep 6, 20575 (2016). https://doi.org/10.1038/srep20575
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20575
This article is cited by
-
Insight into broad substrate specificity and synergistic contribution of a fungal α-glucosidase in Chinese Nong-flavor daqu
Microbial Cell Factories (2023)
-
Identifying carbohydrate-active enzymes of Cutaneotrichosporon oleaginosus using systems biology
Microbial Cell Factories (2021)
-
Novel GANAB variants associated with polycystic liver disease
Orphanet Journal of Rare Diseases (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
