
Abstract
Adenoviral early region 1A (E1A) is a viral gene that can promote cellular proliferation and de-differentiation in mammalian cells, features required for the reprogramming of somatic cells to a pluripotent state. E1A has been shown to interact with OCT4 and as a consequence, to increase OCT4 transcriptional activity. Indeed, E1A and OCT4 are sufficient to revert neuroepithelial hybrids to pluripotency, as demonstrated in previous cell fusion experiments. However, the role that E1A might play in the generation of induced pluripotent stem cells (iPSCs) has not been investigated yet. In this report, we show that E1A can generate iPSCs in combination with OCT4 and KLF4, thus replacing exogenous SOX2. The generated iPSCs are bona fide pluripotent cells as shown by in vitro and in vivo tests. Overall, our study suggests that E1A might replace SOX2 through enhancing OCT4 transcriptional activity at the early stages of reprogramming.
Similar content being viewed by others

Introduction
Somatic cells can acquire a pluripotent fate after ectopic expression of Oct4, Sox2, Klf4, and Myc1, from which Myc can be omitted2. Although OCT4 and SOX2 together with NANOG co-occupy the core targets of the pluripotency transcriptional network in embryonic stem cells (ESCs)3, Nanog is not essential to maintain pluripotency4. In contrast, repression of Oct4 or Sox2 in ESCs promotes differentiation5,6. OCT4 and SOX2 proteins dimerize on DNA to activate their target genes in order to specify pluripotency7. Interestingly, single-point mutations introduced in the SOX2-OCT4 interacting surface are sufficient to prevent their cooperative binding, which in turn precludes the maintenance and induction of pluripotency8,9.
In differentiated cells, OCT4 can activate gene transcription when its DNA binding site is located close to the TATA box. However, OCT4 requires additional factors to induce transcription when the DNA binding sites are distantly located from the TATA element. In fact, a remotely bound OCT4 requires coactivators for bridging the distance between the OCT4 binding site and the TATA box10. The adenovirus early region 1A (E1A) protein can function as such a coactivator and can enhance OCT4 transcriptional activity in somatic cells10. E1A does not directly bind to DNA, instead E1A interacts with components of the general transcriptional machinery such as the TATA-binding proteins and to several transcription factors. Indeed, the conserved domain 3 of the E1A protein has been described to bind to the OCT4 C-terminal domain10. Furthermore, Hamada and colleagues demonstrated that the combined expression of OCT4 and E1A is sufficient to revert neuroepithelial-like cell hybrids to pluripotent embryonic carcinoma-like cells in cell fusion experiments11. Taking together, E1A can enhance OCT4 transactivation activity in somatic cells and both proteins can together reprogram differentiated cells in a cell fusion context. Therefore, we sought to investigate the potential role of E1A in reprogramming somatic cells into induced pluripotent stem cells (iPSCs).
Results
E1A can replace exogenous Sox2 in iPSCs reprogramming
As E1A have been described to reprogram neuroepithelial-like cells into pluripotency11, we assessed whether E1A could also contribute to iPSC formation. Mouse embryonic fibroblasts (MEFs) containing an Oct4-green fluorescent protein (GFP) reporter construct were transduced using several combinations of retroviral-defective particles coding for Oct4 (O), Sox2 (S), Klf4 (K) and E1A (E) (Fig. 1A). MEFs could not be reprogrammed into iPSCs using OK alone, as shown by the lack of Oct4-GFP—positive colonies. However the addition of E1A to OK led to the formation of iPSC colonies (Fig. 1A), indicating that E1A could replace exogenous SOX2. Interestingly, E1A could only substitute for exogenous SOX2 and not for exogenous OCT4 or KLF4 in iPSC induction (Fig. 1B).

E1A can replace exogenous Sox2 in reprogramming MEFs into iPSCs.
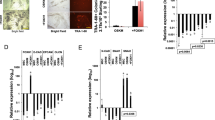
(A) Average number of Oct4-GFP—positive (GFP+) iPSC colonies generated from MEFs after transduction with different combinations of Oct4 (O), Klf4 (K), E1A (E) and Sox2 (S). Colonies were counted at day 22 after viral transduction. Error bars represent standard deviation of the mean of 5 biological replicates. (B) Average number of Oct4-GFP—positive (GFP+) iPSC colonies as described in (A). Colonies were counted at day 19 after viral transduction. Error bars represent standard deviation of the mean of 2 biological replicates. (C) Sox2-null ESCs were transfected with hygromycin-resistant PiggyBac plasmids coding for Sox2, Oct4, or E1A and the empty vector as a negative control. After 1 week in culture in the presence of doxycycline (1 μg/ml) and hygromycin (300 μg/ml), AP staining was performed to quantify the number of surviving ESC colonies and the number of AP—positive colonies per cm2 was counted. The rescue index was calculated by dividing the number of AP—positive colonies for each construct by the number of AP—positive colonies in the empty PiggyBac vector. Error bars represent standard deviation of the mean of 6 biological replicates. (D) 293T cells were transfected with expression vectors encoding the gene of interest coupled to an epitope tag: E1A-FLAG plus Sox2-myc and Oct4-V5. Nanog-FLAG plus Sox2-myc and Oct4-V5 served as a positive control. Immunoprecipitated E1A- or NANOG-FLAG and co-immunoprecipitated SOX2-myc and OCT4-V5 are shown by Western blot. Input corresponds to 2% of the cell lysis extract. (E) The transactivation activity of OCT4 in the presence or absence of E1A was evaluated using the 6wtk luciferase reporter and plotted relative to the levels of the empty vector control. Error bars reflect standard errors (biological replicates: n = 3).
Next, we sought to investigate the role of E1A during reprogramming. To this end, we assessed whether E1A could substitute exogenous SOX2 through the activation of OCT4-SOX2 target genes using Sox2-null ESCs, in which Sox2 is expressed from a transgene that can be repressed upon doxycycline induction5. Hygromycin-resistant PiggyBac vectors coding for SOX2, OCT4, E1A and the empty vector were transfected into Sox2-null ESCs. One week after addition of hygromycin and doxycycline, the rescue index that corresponds to the number of alkaline phosphatase (AP)—positive colonies in the presence of each construct divided by the number of AP—positive colonies in the presence of the empty PiggyBac vector was calculated. Our results show that E1A could not rescue Sox2-null ESC self-renewal (Fig. 1C). Furthermore, Sox2-null ESC lines could be established from Sox2-null ESCs transfected with OCT4 and SOX2 but not with E1A and the empty vector (data not shown). These results demonstrate that E1A does not replace exogenous SOX2 through the activation of the OCT4-SOX2 target genes. We verified OCT4-E1A interaction using a co-immunoprecipitation experiment in which E1A-FLAG was overexpressed in the presence of SOX2-myc and OCT4-V5. NANOG-FLAG was used as a positive control for a double OCT4-SOX2 interaction. After pulling-down with an anti-FLAG antibody, we could demonstrate E1A interaction with OCT4 but not with SOX2 (Fig. 1D). Finally, we assessed whether E1A could enhance OCT4 transcriptional activity using a luciferase assay. A previous report has demonstrated that a certain ratio of OCT4 and E1A is necessary for efficient activation of a luciferase reporter containing POU octamer motifs10, thus we tested three different OCT4:E1A ratios. Our results show an increased transcriptional enhancement by OCT4 in the presence of E1A in all three conditions. Therefore, we conclude that E1A is able to interact with OCT4 and substitute for exogenous SOX2 in iPSC reprogramming, most likely, through the enhancement of OCT4 transcriptional activity10.
iPSCs generated with exogenous E1A are fully pluripotent
Next, we sought to verify the pluripotent potential of MEF-derived iPSCs generated with the novel OKE reprogramming cocktail. Two OKE iPSC clones, OKE-1 and OKE-2, were picked (Fig. 2A), expanded and analyzed. Both clones expressed the Oct4-GFP reporter (Fig. 2B) and stained positive for the pluripotency-markers alkaline phosphatase, OCT4, NANOG and SSEA-1 (Fig. 2C–F). Additionally, endogenous Oct4, Nanog, Sox2, Klf4 and Myc were expressed at levels similar to those of ESCs (Fig. 2G), while the exogenous factors were effectively silenced (Fig. 2H), as shown by quantitative RT-PCR. OKE iPSCs displayed an unmethylated Oct4 promoter, characteristic for pluripotent cells (Fig. 2I). PCR genotyping demonstrated the presence of the OKE transgenes and confirmed the absence of exogenous Sox2 (Fig. 2J). OKE-1 and OKE-2 were propagated in vitro for 30 passages, probing the stability of the new acquired cell fate identity. The differentiation potential of iPSCs in vitro was investigated by embryoid body formation, which was generated using the hanging-drop method. After 2 weeks of differentiation, specific cell types for ectoderm (TUBB3), endoderm (SOX17) and mesoderm (ACTA2) were detected by immunofluorescence microscopy (Fig. 3A). The in vivo differentiation potential was evaluated by teratoma formation. After 6–8 weeks of subcutaneous injection of iPSC lines into nude athymic mice, teratomas containing tissues of all three germ layers were observed (Fig. 3B). Additionally, OKE iPSCs were aggregated with mouse morula-stage embryos and were able to contribute to all germ layers as shown by PCR-genotyping for the GFP transgene (Fig. 3C). Germline contribution was observed by the presence of Oct4-GFP—positive cells in the gonads of 13.5-days post-coitum female and male embryos (Fig. 3D). Taken together, our data indicates that OKE iPSC lines exhibit the capacity to differentiate into cells of all three germ layers. In summary, we generated bona fide iPSCs with the viral factor E1A in combination with Oct4 and Klf4, in the absence of exogenous Sox2.

Molecular characterization of OKE iPSCs.
(A) OKE GFP—positive cells observed 15 days after viral transduction. Scale bars, 100 μm. (B) Oct4-GFP expression in two stable derived OKE cell lines at passage 3. Scale bars, 100 μm. (C) OCT4, (D) AP, (E) NANOG and (F) SSEA-1 immunofluorescence staining images of OKE-1 and -2 clones at passage 5. Scale bars, 100 μm. (G) Expression levels of pluripotency genes measured by qRT-PCR at passage 5. Data were plotted relative to ESCs. Error bars indicate standard deviation of calculations based on ΔΔCt-values obtained from two housekeeping genes, Actb and Rpl37a. (H) Silencing of the retroviral vectors shown by qRT-PCR. OKE-1 and OKE-2 were analyzed at passage 24 and 14, respectively. Data were normalized to Hprt and plotted relative to freshly transduced MEFs. (I) Methylation analysis of the Oct4 promoter in OKE iPSCs passage 5 compared with ESCs and MEFs. Open and filled circles represent unmethylated and methylated CpGs, respectively. X represents an undetermined CpG. (J) PCR genotyping to detect retroviral insertions in OKE-1 and -2 iPSC clones at passage 5. Freshly transduced MEFs and wild-type ESCs were used as a positive and negative control, respectively. As expected, OKE-1 and -2 iPSCs do not contain the Sox2 exogenous transgene.

Differentiation potential of OKE iPSCs.
(A) Immunocytochemistry for marker proteins representative of the three germ layers, TUBB3 (ectoderm), SOX17 (endoderm) and ACTA2 (mesoderm), after in vitro differentiation of OKE iPSCs (passage 10) by embryoid body formation. Scale bars, 150 μm. (B) Teratomas generated by OKE iPSC clones at passage 5 contained tissues of all three germ layers: ectoderm (keratinocyte and neural rosettes), endoderm (gut-like endothelium) and mesoderm (muscle and cartilage). (C) GFP-PCR genotyping to detect contribution of OKE-1 and -2 iPSCs in chimeric animals to organs of all three germ layers: brain (ectoderm), liver (endoderm) and heart (mesoderm). ESCs containing an Oct4-GFP construct and non-transgenic MEFs served as positive and negative controls, respectively. (D) Germline contribution visualized by Oct4-GFP—positive cells in a female and a male 13.5 days post-coitum embryonic gonad.
Discussion
OCT4 and SOX2 constitute the core of the pluripotency network and cooperatively bind and activate their target genes3. Indeed, Sox2-null ESCs differentiate into trophectoderm after SOX2 depletion, demonstrating that SOX2 is required to maintain pluripotency5. Interestingly, Sox2-null differentiation can be rescued upon Oct4 overexpression, suggesting that the main role of SOX2 is to sustain OCT4 expression5. In line with these results, we speculate that E1A can substitute exogenous SOX2 during iPSC reprogramming due to their ability to enhance OCT4 transcriptional activity10. OCT4-rescued Sox2-null ESCs express a subset of Oct-Sox enhancer genes, such as Utf1 or Fgf4, at similar levels than ESCs5. Based on these findings Masui and colleagues concluded that, in the presence of sufficient levels of OCT4, SOX2 is not required to activate a subset of genes containing Oct-Sox elements5, most of which are key genes in the pluripotency network3. E1A and OCT4 alone cannot activate Oct-Sox enhancers (e.g. Fgf4)12, an observation that is also demonstrated by the inability of E1A to rescue Sox2-null ESCs (Fig. 1C). Thus, our results point to E1A’s bridging capacity between OCT4 and the basal transcriptional machinery as the main function underlying its reprogramming potential. However, OKE reprogramming efficiency is much lower than OKS, suggesting that SOX2 can enhance OCT4 transcriptional activity more strongly than E1A or that SOX2 can play other roles that cannot be replaced by E1A.
The molecular events leading to the reprogramming of somatic cells into iPSCs remain unknown. However, the available data supports a model with a stochastic phase followed by a hierarchical phase13. In the initial phase, the exogenous factors are overexpressed at non-physiological levels, a scenario that promotes nonspecific binding to low-affinity binding sites and generates a transient transcriptionally unstable state14. In the final phase, some cells are able to activate endogenous pluripotent genes, inducing a hierarchical cascade of events that finally leads to iPSC generation. In this model, endogenous Sox2 has been proposed to be the upstream gene that activates the endogenous pluripotency program at the late stage of iPSC induction13. In addition, experiments in which each of the four exogenous factors is added sequentially to the cells have demonstrated that the sequence ending with the addition of exogenous Sox2 improves the reprogramming efficiency15. All these evidences point to exogenous SOX2 not being essential to launch the reprogramming program. Thus we hypothesize that reprogramming initiation requires enhanced OCT4 transcriptional activity, which can proceed in the absence of exogenous SOX2, but that reprogramming termination depends on endogenous SOX2. Further studies are required to determine at which time point of the iPSC generation needs endogenous Sox2 to be activated. In summary, our study provides a new insight into the reprogramming mechanism and demonstrates that E1A can replace exogenous SOX2 due to the enhancement of OCT4 transcriptional activity, most likely by augmenting its interaction with the basal transcription machinery.
Methods
Cell Lines
MEFs were derived from mice containing a GFP transgene driven by the Oct4 promoter16. Inducible Sox2-null ESCs carrying the transgenes tet-off Sox2, a tet transactivator and a constitutive red-fluorescent protein transgene (DsRed) have been previously described5. Mouse experiments were performed in accordance with the recommendations of the Federation of Laboratory Animal Science Associations (FELASA) and were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV) of the state of North Rhine-Westphalia (Germany).
Cell Culture Conditions
MEFs were cultured in DMEM medium, supplemented with 10% fetal bovine serum. ESC medium was used for all pluripotent cell lines and consisted of Knockout DMEM supplemented with 20% Knockout serum replacement, 0.2× ß-Mercaptoethanol, 1× nonessential amino acids and 1× penicillin/streptomycin/glutamine and 1,000 U/ml leukemia inhibitory factor (in house preparation).
Generation and characterization of iPSCs
pMX retroviral plasmids coding for Oct4 (Addgene # 13366), Klf4 (Addgene # 13370), Sox2 (Addgene # 13367), Myc (Addgene # 13375)1, E1A (lacking the N-terminus and conserved region 1) (cloned in house) were cotransfected with the pCL-ECO packaging plasmid (Addgene # 12371)17 into 293T cells using Fugene®. Two days later, the supernatants were collected and filtered. MEFs were plated one day before transduction on gelatin-coated 6-well plates (5 × 104 cells/well) and were transduced with 500 μl of Oct4 and Klf4 plus 250 μl of Sox2 or EIA filtered viral-containing supernatants plus 6 μg/ml protamine sulfate. Finally, the supernatant was replaced by ESC medium 48 h after transduction. Each reprogramming experiment was performed at least three times. Alkaline phosphatase staining, immunocytochemistry, methylation analysis, in vitro differentiation, teratoma formation and morula aggregation have been performed as previously described18. Antibodies used for immunocytochemistry are listed in Supporting Information Table S1. Primers are listed in Supporting Information Table S2.
RNA extraction, cDNA synthesis and quantitative real-time PCR (qRT-PCR)
RNA was extracted using the QIAGEN RNeasy Mini Kit (QIAGEN) and cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT-PCR was carried out using iTaq SYBR Green Supermix with ROX (Bio-Rad). Data was plotted using the delta delta Ct algorithm, 2(−∆∆Ct). Primers are listed in Supplementary Table S2.
DNA extraction and genomic PCR
Genomic DNA was extracted using the QIAamp DNA mini kit (QIAGEN). PCR was performed using Taq polymerase (NEB) following the manufacturer’s protocol. Primers are listed in Supplementary Table S2.
Co-immunoprecipitation (Co-IP)
293T cells were transfected with expression vectors encoding C-terminal 3x FLAG-, 3x Myc- or V5-tagged Oct4, Sox2, E1A or Nanog. 24 h after transfection, cells were harvested and lysed in lysis buffer [50 mM Tris-HCl (pH 7.4), 0.1% IGEPAL CA-630, 5 mM EDTA, 150 mM NaCl] followed by a brief sonication using Bioruptor (Diagenode). Lysates were incubated overnight with anti-FLAG antibody bound to Protein G Dynabeads. After washing three times with wash buffer [50 mM Tris-HCl (pH 7.4), 0.1% IGEPAL CA-630, 5 mM EDTA, 125 mM NaCl], immunoprecipitates were eluted by incubation with 200 μg/ml 3× FLAG peptide for 1 h at 4 °C. Immunoprecipitates and whole cell lysates were subjected to SDS-PAGE and immunoblotted with antibodies against FLAG, Myc and V5. Antibodies used for Co-IP are listed in Supplementary Table S1.
Luciferase reporter assay
293T cells were transfected with pGL4.75[hRluc/CMV] together with the reporter constructs 6W-37tk-luc, which drives the luciferase under the control of the canonical binding sequence for POU factors (the ATGCAAAT sequence repeated six consecutive times)19, or with ptk-luc, which was used as a control. The total amount of DNA was maintained constant by adding empty vector. Twenty-four hours after transfection, luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega). The relative luciferase activity (luc/hRluc) was further normalized to that of empty vector control in each reporter.
Sox2-null ESCs self-renewal assay
Sox2, Oct4 and E1A were cloned in the hygromycin-resistant PiggyBac plasmid pPBCAG-cHA-IH, a modification of the previously reported pGG131 plasmid that contains a hygromycin selection cassette20. 450 ng of the PiggyBac coding for Sox2, Oct4 and E1A together with 150 ng of pCAG-PBase plasmid were co-transfected into 5 × 104 Sox2-null ESCs5 using Lipofectamine 2000. Doxycycline (1 μg/ml) was added to the medium 5 h after transfection. Next, cells were transferred onto feeder cells and selected with 300 μg/ml hygromycin for 1 week. Finally, AP was performed to quantify the number of surviving ESC colonies.
Additional Information
How to cite this article: Marthaler, A. G. et al. Enhanced OCT4 transcriptional activity substitutes for exogenous SOX2 in cellular reprogramming. Sci. Rep. 6, 19415; doi: 10.1038/srep19415 (2016).
References
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Nakagawa, M. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26, 101–106 (2008).
Boyer, L. A. et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 (2005).
Carter, A. C., Davis-Dusenbery, B. N., Koszka, K., Ichida, J. K. & Eggan, K. Nanog-Independent Reprogramming to iPSCs with Canonical Factors. Stem cell reports 2, 119–126 (2014).
Masui, S. et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature cell biology 9, 625–635 (2007).
Niwa, H., Miyazaki, J. & Smith, A. G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nature genetics 24, 372–376 (2000).
Remenyi, A. et al. Crystal structure of a POU/HMG/DNA ternary complex suggests differential assembly of Oct4 and Sox2 on two enhancers. Genes Dev 17, 2048–2059 (2003).
Jauch, R. et al. Conversion of Sox17 into a pluripotency reprogramming factor by reengineering its association with Oct4 on DNA. Stem Cells 29, 940–951 (2011).
Tapia, N. et al. Dissecting the role of distinct OCT4-SOX2 heterodimer configurations in pluripotency. Scientific Reports 5, 13533 (2015).
Scholer, H. R., Ciesiolka, T. & Gruss, P. A nexus between Oct-4 and E1A: implications for gene regulation in embryonic stem cells. Cell 66, 291–304 (1991).
Shimazaki, T. et al. Hybrid cell extinction and re-expression of Oct-3 function correlates with differentiation potential. The EMBO journal 12, 4489–4498 (1993).
Ambrosetti, D. C., Basilico, C. & Dailey, L. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Molecular and cellular biology 17, 6321–6329 (1997).
Buganim, Y. et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222 (2012).
Tiemann, U. et al. Counteracting Activities of OCT4 and KLF4 during Reprogramming to Pluripotency. Stem cell reports 2, 351–365 (2014).
Liu, X. et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nature cell biology 15, 829–838 (2013).
Yoshimizu, T. et al. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Development, growth & differentiation 41, 675–684 (1999).
Naviaux, R. K., Costanzi, E., Haas, M. & Verma, I. M. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. Journal of virology 70, 5701–5705 (1996).
Marthaler, A. G. et al. Reprogramming to pluripotency through a somatic stem cell intermediate. PloS one 8, e85138 (2013).
Scholer, H. R., Balling, R., Hatzopoulos, A. K., Suzuki, N. & Gruss, P. Octamer binding proteins confer transcriptional activity in early mouse embryogenesis. The EMBO journal 8, 2551–2557 (1989).
Guo, G. et al. Klf4 reverts developmentally programmed restriction of ground state pluripotency. Development 136, 1063–1069 (2009).
Acknowledgements
We would like to thank Bärbel Schäfer, David Obridge and Sandra Heising for technical assistance. The E1A cloning plasmid was a gift from Tony Kouzarides. The Max Planck Society is funding H.R.S group, whereas the North-Rhine Westphalia Ministry of Innovation, Science and Research as well as the Heinrich Heine University Düsseldorf are funding N.T. Junior Research Group.
Author information
Authors and Affiliations
Contributions
A.G.M. collection and assembly of data, data analysis and interpretation. K.A., U.T., G.W., D.S. and S.V. collection and assembly of data. I.K. provision of study material. H.R.S. designed the concept of the experiment and wrote the manuscript. N.T. designed the concept of the experiment, collection and assembly of data, data analysis and interpretation and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Marthaler, A., Adachi, K., Tiemann, U. et al. Enhanced OCT4 transcriptional activity substitutes for exogenous SOX2 in cellular reprogramming. Sci Rep 6, 19415 (2016). https://doi.org/10.1038/srep19415
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19415
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
