
Abstract
VP40 is one of eight proteins encoded by the Ebola Virus (EBOV) and serves as the primary matrix protein, forming virus like particles (VLPs) from mammalian cells without the need for other EBOV proteins. While VP40 is required for viral assembly and budding from host cells during infection, the mechanisms that target VP40 to the plasma membrane are not well understood. Phosphatidylserine is required for VP40 plasma membrane binding, VP40 hexamer formation and VLP egress, However, PS also becomes exposed on the outer membrane leaflet at sites of VP40 budding, raising the question of how VP40 maintains an interaction with the plasma membrane inner leaflet when PS is flipped to the opposite side. To address this question, cellular and in vitro assays were employed to determine if phosphoinositides are important for efficient VP40 localization to the plasma membrane. Cellular studies demonstrated that PI(4,5)P2 was an important component of VP40 assembly at the plasma membrane and subsequent virus like particle formation. Additionally, PI(4,5)P2 was required for formation of extensive oligomers of VP40, suggesting PS and PI(4,5)P2 have different roles in VP40 assembly where PS regulates formation of hexamers from VP40 dimers and PI(4,5)P2 stabilizes and/or induces extensive VP40 oligomerization at the plasma membrane.
Similar content being viewed by others


Introduction
The Ebola Virus (EBOV) is a lipid enveloped filamentous virus that results in severe hemorrhagic fever in human and non-human primates with up to 90% fatality1. EBOV harbors a negative sense RNA genome that only encodes seven genes, one of which is for the viral matrix protein, VP40. VP40 is the most abundant protein in EBOV virions and can form authentic looking virus like particles (VLPs) that bud from the plasma membrane of live cells in the absence of other EBOV proteins2,3. VP40 is required for the virus to exit from the host cell membrane, suggesting it is a potential drug target2,4,5. In addition to forming the matrix layer in the viral particles, VP40 interacts with several host proteins during trafficking to and while residing at the plasma membrane6,7,8,9,10,11. VP40, which was shown to be a dimer, can also adopt several structural conformations during the viral life cycle including a hexameric filament required for budding12,13,14.
VP40 is a peripheral protein containing 326 amino acids with a N-terminal domain (NTD) that mediates dimerization12 and oligomerization12,15,16 and C-terminal domain (CTD) that mediates membrane binding12,17,18. Several studies have shown that VP40 requires both its NTD and CTD, for association with the PM19. The association with the plasma membrane anionic interface has been proposed to trigger VP40 structural reorganization into a zigzag hexamer12 and subsequently a highly ordered matrix structure that coats the inside of the viral particle17,18,19,20,21.
VP40 interactions with the plasma membrane are mediated by electrostatic interactions between a CTD basic patch and anionic lipids enriched in the plasma membrane inner leaflet12. Additionally, hydrophobic interactions are likely an important part of VP40 plasma membrane assembly, as a hydrophobic loop in the CTD has been found to insert into lipid vesicles17,18. More than a decade ago, VP40 was shown to associate with phosphatidylserine (PS) containing vesicles22,23 and more recently it was shown that PS was essential for VP40 plasma membrane localization and formation of VLPs24. PS is also required for the formation of hexamers in live cells from VP40 dimers. In addition, PS becomes exposed on the outer leaflet of non-apoptotic cells at sites of VP40 budding. The PS exposure is most abundant on VLPs; this observation was expected as PS exposure expedites entry into host immune cells through interaction with the PS binding TIM-1 receptor25,26,27. This finding, however, raised some questions regarding how VP40 maintains the structural integrity of the viral particle since the VP40 interaction partner PS becomes exposed on the exoplasmic leaflet of VLPs. This lead us to hypothesize that other lipids at the plasma membrane may be involved in coordinating and maintaining the VP40-plasma membrane interactions.
Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) is this most abundant phosphoinositide (PIP) in mammalian cells comprising up to 2–5% of the plasma membrane28,29,30. The subcellular location of PI(4,5)P2 is restricted to the plasma membrane inner leaflet (major)28,31,32 and within the nucleus (minor)33,34,35 of mammalian cells. Several viral matrix proteins including the matrix proteins from human immunodeficiency virus type-1 (HIV-1)36, HIV-237 and Mason-Pfizer monkey virus (M-PMV)38,39 require PI(4,5)P2 for association with the PM, matrix assembly and egress. Each of these matrix proteins utilize a basic patch of residues that coordinate the head group of PI(4,5)P2. VP40 also has a highly basic region in its CTD that may interact with PS, PI(4,5)P2, or other PIPs at the plasma membrane. To test this possibility, we employed cellular and in vitro assays to examine VP40 association with plasma membrane lipids. Results demonstrate that VP40 associates with PI(4,5)P2 in the plasma membrane and in vitro vesicles and this interaction is a prerequisite for extensive VP40 oligomerization at the plasma membrane and VLP formation.
Results
The VP40-Plasma Membrane interaction is beyond nonspecific electrostatics
An EGFP fusion tagged version of VP40 localized to the inner leaflet of the plasma membrane of COS-7 cells where VLPs are formed. VLP formation at the plasma membrane was observed as early as 8 hours post transfection in COS-7 cells and were abundant at 14 hours post transfection (Fig. 1A). To determine the average cellular distribution and plasma membrane localization of VP40 and control proteins, PLCδ-PH (PI(4,5)P2 biosensor)40, Lact C2 (PS biosensor)41 and HIV-1 Gag were each transfected into COS-7 cells and imaged 14 hours post transfection with a Zeiss LSM 710 confocal laser scanning microscope (Fig. 1A). Previous work observed no significant changes in VP40 PM localization when the PM was neutralized with sphingosine, a membrane permeant base41. To verify VP40 exhibited similar behavior in COS-7 cells, we repeated these experiments using 75 μM sphingosine or an equivalent volume of vehicle (ethanol). Sphingosine or vehicle were added to the media of cells expressing the polybasic charge sensor KRΦ42 or VP40 transfected cells for 1 or 12 hours. As previously reported24, plasma membrane localization of KRΦ was significantly reduced upon sphingosine treatment, compared to vehicle (Fig. 1B,C) but no significant decrease in VP40 plasma membrane localization was observed with either sphingosine treatment or vehicle control (Fig. 1B,C). These findings indicated that VP40 localized to the PM through a mechanism independent of nonspecific electrostatics in COS-7 cells.

VP40 localizes to the plasma membrane of COS-7 cells.
(A) Representative images of PLCδPH-EGFP, LactC2-EGFP, EGFP-VP40 and HIV-GAG-EGFP 14 hours post transfection in COS-7 cells. (B) Representative images of KRΦ-mRFP and EGFP-VP40 treated with either vehicle (EtOH 1:500) or 75 uM sphingosine for 1 or 14 hours. (C) Percent plasma membrane localization of KRΦ-mRFP and EGFP-VP40 following one and 12 hour 75 uM sphingosine treatments. All data was quantified in MATLAB where %PM localization was defined by (PM intensity/total intensity)*100. Data was normalized to the %PM localization of each construct with the 1 or 12 hour vehicle treatment, which was set to 100%. Error bars are ± SEM from at least 20 cells over three independent experiments. All scale bars are 10 μm.
Competition assays to identify potential VP40-lipid interaction partners
To identify potential anionic lipid substrates in addition to PS at the plasma membrane, EGFP-VP40 (0.4 ug DNA) was co-transfected with a low level (0.15 ug DNA) or excess level (0.5 ug DNA) of a lipid biosensor with a mCherry or mRFP tag. This allowed for competition of various lipids including PI(4,5)P2 (PLCδ-PH), PI(4)P (SidM-P4M), PI(3)P (EEA1), PI(3,4,5)P3 (AKT-PH) and PS (LactC2). Polybasic charge sensors KRΦ (peripheral) and R-Pre (lipidated) were used to mask anionic lipids as a positive control and mCherry alone was used as a negative control. The quantified plasma membrane localization of VP40 and control proteins with low or excess of competitor protein are shown in Fig. 2. VP40 plasma membrane localization was significantly reduced compared to VP40 alone when co-transfected with the PLCδ-PH PI(4,5)P2 biosensor, Akt-PH PI(3,4,5)P3 biosensor, excess SidM PI(4)P biosensor SidM, excess PI(3)P biosensor EEA1, excess PS biosensor LactC2 and polybasic charge sensors KRΦ and R-Pre. VP40 localization was not significantly decreased when co-transfected with mCherry. Representative images from each assay are shown in supplementary Figure 1. The polybasic charge sensors also produced a significant decrease in VP40 and LactC2 PM localization indicating that the loss in VP40 PM localization is likely due to masking of PS at the PM. Together these findings suggested that VP40 may likely require phosphoinositide (PIP) substrates at the PM in addition to PS. To further test this hypothesis, lipid phosphatase enzymes were co-transfected with VP40 to assess the role of some PIPs in VP40 localization.

VP40 plasma membrane localization is disturbed by overexpression of various lipid biosensors.
(A) Percent plasma membrane localization of PLCδ-PH-EGFP construct (0.4 μg) when co-transfected with low levels (0.15 μg) and excess (0.5 μg) competitor protein. (B) Percent plasma membrane localization of LactC2-EGFP construct when co-transfected with low levels and excess competitor protein. (C) Percent plasma membrane localization of EGFP-VP40 construct when co-transfected with low levels and excess competitor protein. (D) Percent plasma membrane localization of HIV-GAG-EGFP construct when co-transfected with low levels and excess competitor protein. Each data set represents the average percent plasma membrane localization of at least 15 independent cells from three independent experiments. All data was quantified in MATLAB where %PM localization is defined by (PM intensity/total intensity)*100. Data was normalized to the %PM localization of each construct when transfected alone in COS-7 cells for the same time (14 hours). Error bars are ± SEM. Significant changes (p < 0.05) from normal plasma membrane localization are marked with a star (*).
Constitutive depletion of PI(4,5)P2 in Live Cells
Consitutive enzyme Myc-5’PtaseIV were used to deplete PI(4,5)P2. A constitutive 5-phosphatase Myc-5′PtaseIV-WT (active) and Myc-5′PtaseIV-Δ1 (inactive) have been effective tools to investigate the PI(4,5)P2 binding properties of various proteins including, HIV-1 Gag36. This phosphatase construct is myristoylated post transnationally, which targets it to the plasma membrane. When VP40 was co-transfected with Myc-5′PtaseIV-WT, plasma membrane localization was significantly decreased similar to the response of PLCδ PH in the presence of Myc-5′PtaseIV-WT (Fig. 3A,B). Under similar conditions the HIV-1 GAG exhibited remarkable depletion of PM localization while the PS biosensor Lact C2 did not. In contrast, VP40 PM localization was not significantly reduced in the presence of a the catalytically inactive Myc-5′PtaseIV-Δ1.

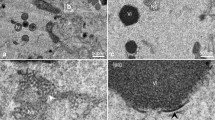
Constitutive enzyme depletion of lipid substrates results in decreased VP40 VLP formation.
(A) Representative images of EGFP constructs PLCδ-PH-EGFP (N = 20), LactC2-EGFP (N = 20), EGFP-VP40 (N = 24) and HIV-GAG-EGFP (N = 20) co-transfected with Myc-5’PtaseIV-WT, or Myc-5’PtaseIV-Δ1 in COS-7 cells. All scale bars are 10 μm. N indicates the numbers of cells imaged over three independent transfections. (B) Quantitative analysis of plasma membrane localization of each construct under each co-transfection condition. All values represent at least 20 independent cells over three independent experiments ± SEM. All data was quantified in MATLAB where %PM localization is defined by (PM intensity/total intensity)*100. Data was normalized to the %PM localization of each construct when transfected alone in COS-7 cells for the same time (14 hours). Significant changes (p < 0.05) from normal PM localization are marked with a star (*). (C) A VLP egress assay was performed. VLPs were collected and purified from EGFP-VP40 transfected cells (see methods) and the EGFP content was quantified using a GFP ELISA kit (Cell Biolabs) and the percentage VLP egress was determined as the ratio of GFP in the VLP sample to the total GFP (VLP+cell lysate) sample. Significant changes (p < 0.05) from normal VLP egress are marked with a star. (D) Scanning electron microscopy (SEM) of COS-7 cells transfected for 14 hours with VP40, EGFP-VP40 +Myc5’PtaseIV-WT, EGFP-VP40 +Myc-5’PtaseIV-Δ1, or EGFP. Images are shown for each sample, two replicates were performed.
To determine if plasma membrane PI(4,5)P2 levels were important for VP40 VLP formation, a VLP egress assay was performed 24 hours post transfection when VP40 was co-expressed with either the active or catalytically inactive 5′-PtaseIV. After collecting VLP and cell lysate (CL) samples (see methods section) an ELISA assay (Cell Biolabs) was used to quantify the percent GFP in each sample. A summary of findings are presented in Fig. 3C where VLP egress of EGFP-VP40 was normalized to 100%. This data is in line with the findings from the confocal imaging experiment where PI(4,5)P2 was essential for normal VLP formation (Fig. 3C). The residual plasma membrane localization and VLP egress could likely be due to incomplete co-transfection of enzymatic constructs with VP40. To determine if morphological changes existed for VLP formation and to further confirm the importance of PI(4,5)P2 in VLP formation, scanning electron microscopy was used to visualize the plasma membrane morphology of cells transfected with EGFP-VP40, EGFP alone, or EGFP-VP40 co-expressed with either Myc5’PtaseIV-WT or Myc5’PtaseIV-Δ1 (Fig. 3D). An abundance of VLPs and smaller bud structures were observed on EGFP-VP40 and VP40 + Myc5’PtaseIV-Δ1 transfected cells. VLPs were not readily observed on VP40 + Myc5’PtaseIV-WT cells, however, smaller bud structures were somewhat visible. EGFP transfected cells did not have significant bud structures but had a visible low abundance of filopodia structures on some cells, which is also observed on some EGFP transfected cells using confocal microscopy (data not shown).
Pharmacological treatments support VP40 phosphoinositide dependence at the plasma membrane
To remove various phospholipid substrates without co-transfection, ionomycin, phenyl arsine oxide (PAO) and wortmannin treatments were used. Ionomycin is a calcium ionophore, where increased cellular calcium activates PLC and induces activation of a lipid scramblase that induces some loss of plasma membrane asymmetry. As a result, the PI(4,5)P2 and PI(4)P head groups are hydrolyzed by PLC and PS becomes more exposed on the outer surface of the plasma membrane, as a function of scramblase activity43. Representative images of VP40 pre-treatment and post-treatment with ionomycin demonstrate VP40 dissociation from the plasma membrane as well as an increase in PS exposure as detected with the extracellular PS biosensor Annexin V (Figs 4A and S2C), The average %PM localization of each construct tested was quantified versus time (Fig. 4B). It was previously reported that VP40 expression in mammalian cells induces PS exposure on the exterior of VLPs in HEK293 cells24; this was also observed in EGFP-VP40 transfected COS7 cells before ionomycin treatment (Fig. 4C). Furthermore, VP40 dissociated from the plasma membrane upon ionomycin treatment similarly to PI(4,5)P2 and PI(4)P biosensors, PLCδ-PH and SidM-P4M, respectively (Fig. 4B). Images of control biosensors are shown in Figure S2.

PIP depleting pharmacological treatments yield a decrease in VP40 PM association.
(A) Representative images of VP40 transfected into COS-7 cells for 14 hours pre- and 5 minutes post 10 μM ionomycin treatment in 1x Annexin V binding buffer. Annexin V APC was added five minutes post treatment (1:25) and imaged after a five-minute incubation. Scale bars are 10 μm. (B) Percent plasma membrane localization of each construct was quantified over a 5 minute 10 μM ionomycin treatment, where data were normalized to pre-treatment plasma membrane localization of 100%. (C) Representative images of EGFP-VP40 pre-ionomycin treatment with AnnexinV-APC staining are shown to demonstrate PS exposure in VP40 expressing cells prior to ionomycin addition. (D) Representative images of EGFP-VP40 (N = 22), AKT-PH-mRFP (N = 34), LactC2-EGFP (N = 16), PLCδ-PH-EGFP (N = 16) and SidM-P4M-GFP (N = 24) with 30 minute, 10 μM PAO treatment or equal volume DMSO control treatment. Scale bars are 10 μm. (E) Percent PM localization of PAO treated cells compared to DMSO treated cells, normalized to 100%. (F) Representative images of EGFP-VP40 (N = 20), AKT-PH (N = 18), PLCδ-PH-EGFP (N = 18), SidM-P4M-GFP (N = 22) and EEA1-YFP, with 1 hour, 100 nM Wortmannin treatment or equal volume DMSO control treatment. (G) Percent PM localization of Wortmannin treated cells compared to DMSO treated cells, normalized to 100%. Average values ± SEM are shown for each construct, N indicates the numbers of cells imaged over three independent transfections. Significance was determined with a two-tailed T-test, p values less than 0.05 are marked with a star.
PAO, a PI4 kinase inhibitor resulted in a significant decrease in PI(4)P and PI(4,5)P2 at the PM as shown with biosensors SidM-P4M and PLCδ-PH (Fig. 4D). Additionally, VP40 PM localization was significantly reduced with PAO treatment versus the DMSO control (Fig. 4D,E). The PI3 kinase inhibitor, Wortmannin, resulted in a significant decrease in AKT-PH plasma membrane localization but had no significant effect on VP40, compared to the DMSO control (Fig. 4F,G). Additionally, the PI(3)P biosensor EEA1-FYVE was displaced from endosomes following Wortmannin treatment but not vehicle control (Fig. 4F). Together, the pharmacological treatments reinforce the hypothesis that phosphoinositides promote VP40 localization at the plasma membrane. However, each of the treatments used have promiscuity in their substrate selectivity and therefore, a more specific lipid depletion method was used to confirm results.
Real time lipid depletion and lipid binding assays reveal VP40 cellular and in vitro phosphoinositide selectivity
A rapamycin inducible lipid depletion system generated by Hammond et al. was used to specifically deplete PI(4,5)P2 and PI(4)P in real time44. VP40 or control proteins (LactC2 or PLCδ-PH) were co-transfected with a plasma membrane anchored Lyn11-FRB-CFP and one of four pseudojanin (PJ)-FKBP cytosolic constructs. Four PJ-constructs were used to deplete PI(4,5)P2 (PJ-INPP5E), PI(4)P (PJ-Sac1), PI(4,5)P2 and PI(4)P (PJ-WT), or neither (PJ-Dead, inactive)44. Representative images of the EGFP construct before and after each experiment are shown in Fig. 5A. The percent plasma membrane localization was calculated before rapamycin addition (set to 100%) and after rapamycin addition, the % PM localization post depletion is shown in Fig. 5B. VP40 had a slight decrease in PM localization when PI(4,5)P2 or PI(4)P was depleted, but it wasn’t statistically significant (with PJ-WT, PJ-INPP5E and PJ-Sac1) compared to the inactive construct (PJ-Dead) (Fig. 5B). The change in VP40 PM localization was much less than the control PLCδ-PH construct. The molecular basis of this finding is unknown, further studies will be required to fully determine the mechanistic differences. Further experiments as described below suggest PI(4,5)P2 is a critical determinant of extensive VP40 oligomerization. Additionally, the VLPs present on cells co-transfected with Lyn11 are not as prominent as with non-PM constructs (Fig. 2, Figure S1). When VP40 was co-transfected with Lyn11, PM localization was significantly reduced compared to VP40 expression without Lyn11 (Figure S3A, S3B).

Real time lipid depletion and in vitro binding reveal VP40 membrane association is significantly enhanced by PI(4,5)P2.
(A) Representative images of PLCδ-PH-EGFP, LactC2-EGFP and EGPF-VP40 pre and post rapamycin treatment with PJ-WT-FKBP-mRFP, PJ-INPP5E-FKBP-mRFP, PJ-Sac1-FKBP-mRFP and PJ-Dead-FKBP-mRFP phosphatase constructs. PM anchor Lyn11-FRB-CFP was used in each experiment. At least 3 independent experiments were used for each condition. (B) % PM localization values were plotted for PLCδ-PH-EGFP, LactC2-EGFP and EGFP-VP40 with depletion by PJ-WT-FKBP-mRFP, PJ-INPP5E-FKBP-mRFP, PJ-Sac1-FKBP-mRFP and PJ-Dead-FKBP-mRFP phosphatase constructs. % PM Localization was calculated using MATLAB for each cell post rapamycin addition, normalized to pre rapamycin addition. Average values ± SEM are shown for each construct. Significant changes (p < 0.05) for each PJ-construct as compared to those with the PJ-Dead construct are marked with a star. (C) Representative SDS PAGE gel images of VP40 in the supernatant (SN) and pellet (P) with each liposome condition, 4 replicates are shown for each condition. (D) Average fraction VP40 bound to multilamellar vesicles of various composition with average values plotted ± SEM. Significant changes (p < 0.05) are marked with a star.
A multilamellar vesicle (MLV) sedimentation assay was used to determine if VP40 binds to PI(4,5)P2 and other PIPs in vitro. Representative gel images are shown in Fig. 5C and quantified results are shown in Fig. 5D. There is a significant increase in VP40 MLV binding to vesicles containing both PS and either PI(3)P, PI(4,5)P2, or PI(3,4,5)P3 compared to vesicles harboring PIPs in the absence of PS. Notably, VP40 displayed a ~3-fold increase in binding to PI(4,5)P2 containing vesicles when PS was present. VP40 also associated with PI(3)P and PI(3,4,5)P3 containing vesicles in this manner but not those containing PI(4)P. While VP40 associates with PI(3)P and PI(3,4,5)P3 in vitro, PI(3)P is generally found on vesicles during endocytosis31 and PI(3,4,5)P3 represents only 2–5% of PI(4,5)2 levels at the plasma membrane28,45. Therefore, the combination of cellular and in vitro data suggest VP40 association with PI(4,5)P2 may be the most important PIP interaction for plasma membrane assembly. Nonetheless, interactions with or the importance of other PIPs cannot be discounted for VP40 as PI(4)P and PI(3,4,5)P3 play some role in PM localization as evidenced by the competition assay and other PIPs (PI(3)P, PI(4)P, or PI(3,5)P2) could play some sort of trafficking role in VP40 targeting to the PM.
PI(4,5)P2 is required for VP40 oligomer formation at the plasma membrane
Adu-Gyamfi et al. recently demonstrated that PS was required for VP40 oligomerization and VLP formation as PS depleted cells VP40 had a low level of oligomerization compared to the controls25. Moreover, the inhibition of VP40 oligomerization was quite apparent as Number and Brightness analysis (N&B) revealed PS was essential for significant oligomerization to occur beyond the constitutive VP40 dimer. To determine the role of PI(4,5)P2 in VP40 oligomerization, raster image correlation spectroscopy (RICS) and N&B analysis were used to investigate the diffusion coefficient and average oligomer size of VP40 at the plasma membrane with or without PI(4,5)P2 depletion.
The diffusion coefficient of EGFP-VP40 was determined for each sample at a region near the surface of each cell using an Olympus FV1000 2-photon confocal microscope. For each cell, a stack of 100 images was collected according to specifications in the methods section. Data analysis in SimFCS was then performed to determine the diffusion coefficient and the oligomerization state of EGFP-VP40 or EGFP. The diffusion coefficient of EGFP-VP40 and EGFP-VP40 co-transfected with the catalytically inactive Myc-5’PtaseIV construct had no significant differences (Fig. 6A). However, when VP40 was co-transfected with the WT phosphatase Myc-5’PtaseIV, the diffusion coefficient of EGFP-VP40 was significantly increased compared to the diffusion coefficient of EGFP-VP40 with the inactive phosphatase (Fig. 6A).

VP40 diffusion and oligomerization near the plasma membrane of the COS-7 cells were modified after PIP depletion.
(A) Diffusion coefficients for EGFP (N = 4), EGFP-VP40 (N = 9), EGFP-VP40+Myc-5’PtaseIV-Δ1 (N = 4) and EGFP-VP40+Myc-5′PtaseIV-WT (N = 8) were calculated using raster image correlation spectroscopy (RICS) in SimFCS. (B) Representative images from EGFP-VP40 (N = 14), EGFP-VP40 + Myc-5 ‘PtaseIV-WT (N = 11) and EGFP-VP40 +Myc-5′PtaseIV- Δ1 (N = 11) number and brightness workflow in SimFCS. The original intensity image is shown in panel 1, the number verses intensity plot in panel 2 and the selection plot of the cell image in panel 3. (C) The percent pixels with a brightness value of 1–1.5 (monomer-hexamer), 1.5–2.0 (hexamer to 12mer) and 2.0 ~ 6.5 (12mer to ~36mer) were calculated per cell and the average value for construct was plotted. Average values shown for all graphs ±SEM. Significant changes (p < 0.05) are marked with a star.
Number and Brightness analysis (N&B) is a method that measures the variance in intensity at each pixel over time (100 image stack) to determine the intensity and brightness of the fluorescent protein at each pixel throughout the image46. As a result, the intensity at each pixel is distinguished from the oligomerization state at each pixel and the results are shown in a brightness (B) vs. intensity plot as shown in Fig. 6B, panel 2. EGFP was used to determine the true brightness of a monomer in COS-7 cells; a multiple of this value indicates an increase in protein oligomerization. The pixels correlating to monomer-hexamer, hexamer-12mer and 12mer and larger were calculated and average (see Equation 1). An image representation of this is shown in Fig. 6B, panel 3. The percentage of pixels in each bin were averaged and plotted for EGFP-VP40, EGFP-VP40 + Myc5’PtaseIV-WT and EGFP-VP40 + Myc5’PtaseIV-Δ1 (Fig. 6C). VP40 co-transfected with WT Myc-5’PtaseIV exhibited a significant decrease in the percentage of 12mer or greater near the surface of the cells and an increase in monomer-hexamer, compared to EGFP-VP40 alone. There was no significant difference in oligomerization between EGFP-VP40 and EGFP-VP40 + Myc5’PtaseIV-Δ1 (Fig. 6C). These data indicate that PI(4,5)P2 is an important component of stabilization of VP40 filaments at the plasma membrane surface at, which is likely a critical step in forming the extensive VP40 matrix layer that completely underlies the viral lipid envelope.
Discussion
VP40 is the most abundant protein in EBOV virions and is required for viral particle formation and egress. Because of its critical role in the life cycle of the virus, VP40 has been suggested as a potential therapeutic target4,47. More than a decade ago, VP40 was shown to associate with lipid vesicles containing PS22,23 as well as lipid raft regions of the plasma membrane48. Additionally, VP40 is able to associate with PS containing vesicles in the absence of phosphoinositides, a necessary step in promoting VP40 oligomerization of VP40 dimers22,23,24. However, until recently, mechanistic information of VP40 interactions with lipids was limited. First, VP40 was shown to interact with the hydrocarbon core of lipid membranes containing PS and this interaction was shown to be important for VLP formation17,18. Recently, cellular studies by Adu-Gyamfi et al. demonstrated that cellular PS is essential for VP40 plasma membrane localization, oligomerization and VLP egress24. However, it was unclear whether VP40 required additional lipid substrates to maintain the structural integrity of the virus particle when PS was exposed to the extracellular side of the VLP.
Several viral matrix proteins selectively bind PI(4,5)P2 at the PM36,37,49. This phosphoinositide is enriched at the PM, making up over 99% of the bis-phosphorylated species29,50. While VP40 can associate with PS containing membranes lacking PI(4,5)P2, we have shown that this lipid is required for normal VP40 localization to the plasma membrane and large oligomer formation in COS-7 cells (Fig. 2C, Supplementary Figure 1, Figure 3, Figure 4, Figure 5). The loss of plasma membrane localization was not simply due to a decrease in the overall membrane anionic charge; VP40 plasma membrane localization was not decreased when the plasma membrane was neutralized (Fig. 1B,C). When KR-Φ or R-Pre were over expressed with VP40, there was significantly less VP40 at the PM, likely because PS was masked by the polybasic probes as LactC2 PM localization was also reduced (Fig. 2B,C, Supplemental Figure 1).
MLV binding analysis indicated that VP40 associates with PI(4,5)P2 as well as PI(3)P and PIP3 (Fig. 5C,D). These interactions were weak in the absence of PS in the MLVs but significantly increased when PS was present. In contrast, binding to PI(4)P was not evident indicating VP40 may harbor some PIP binding selectivity. While VP40 binds PI(3)P containing vesicles in vitro, PI(3)P may not have a direct role in VP40 plasma membrane localization (Fig. 4F,G). From the combination of cellular and in vitro work, the current data suggests that PI(4,5)P2 is important for VP40 plasma membrane localization, extensive oligomerization and stabilization of VP40 oligomers at the plasma membrane surface, as well as VLP egress. Interestingly, PS was previously shown to be critical for forming oligomers larger than the dimer24 and here we find that PI(4,5)P2 was critical for oligomers 12mer and greater but not likely those between a dimer and 12mer. While additional investigation is required to fully resolve how PS and PI(4,5)P2 structurally act as molecular switches in VP40 oligomerization, it seems these lipids have distinct roles in VP40 assembly. We propose PS is essential for the initial association of VP40 with the plasma membrane and subsequent oligomerization of the dimer. As PS exposure increases on the outer leaflet of the plasma membrane, we propose PI(4,5)P2 is important for stabilizing the extensive VP40 oligomers that form and may be a key regulator of VP40 concatenation to form the underlying protein matrix layer.
VP40 inserts a CTD hydrophobic loop into the plasma membrane upon association17,18; this increases residence time of lipid binding and is likely an essential step in structural rearrangement of VP40 oligomers. It is unknown if this event is induced or strengthened by a specific lipid substrate or if PS binding to VP40 is a prerequisite for PI(4,5)P2 binding and oligomerization. HIV-1 Gag proteins harbor a myristate required for membrane retention51 and also associate with PI(4,5)P2, which is required for assembly and budding36,52,53. Further, Gag multimerization can increase membrane affinity54 while membrane association of Murine Leukemia Virus Gag is greatly enhanced with increasing PS49. Although VP40 is not known to have a lipidation site, the CTD hydrophobic loop insertion17,18 could require a specific lipid trigger for exposure and membrane association. This mechanism of insertion may also help to explain the increased membrane retention of VP40 compared to PLCδ-PH in the rapamycin induced PIP depletion experiments. PLCδ-PH interactions with the plasma membrane surface are primarily through interactions with the anionic surface containing PI(4,5)P2. We hypothesize that in the absence of PI(4,5)P2 VP40 would still be able to interact with PS, harbor hydrophobic interactions with the lipid acyl chains and may have higher avidity due to its oligomerization (hexamer to 12mer).
The differential roles of PS and PI(4,5)P2 in localization to the plasma membrane and oligomerization have yet to be fully elucidated during VP40 trafficking, plasma membrane association and egress. In 2013, Bornholdt et al. identified a cationic patch on the CTD enriched in lysine residues that was required for plasma membrane association12. Additional investigation of the residues within the cationic patch may clarify which residues are important for PS, PI(4,5)P2, or association with other PIPs. Several retroviruses also interact with PS and PI(4,5)P2, but the PS binding site can be blocked by RNA association55,56, which has been proposed to allow proper and specific targeting to the plasma membrane as PS is found on the cytoplasmic face of endomembranes57,58. If such a mechanism exists for VP40 due to the potential of its basic patch to associate with RNA is still unknown.
VP40 interactions with other PIPs in the life cycle of the virus cannot be discounted. In our in vitro assays we observed VP40 to associate with PI(3)P and PI(3,4,5)P3 when PS was present, in a similar fashion to that of PI(4,5)P2. Additionally, competition assays demonstrate blocking access to PI(4)P and PI(3,4,5)P3 also lowered VP40 plasma membrane localization. PI(4,5)P2 levels were clearly important for plasma membrane localization of VP40 as well as VLP egress, which is likely attributed to the role of PI(4,5)P2 in stabilizing the extensive VP40 oligomers that form at the plasma membrane. Nonetheless, PI(4)P and PI(3,4,5)P3 at the plasma membrane could also play a role in stabilization of VP40 oligomers and VP40 membrane retention time.
EBOV or VP40 could also alter cellular lipid metabolism in its favor by regulating a lipid kinase (e.g., PI3K) or inhibition of a lipid phosphatase (e.g., PTEN). Increases in respective PIPs could then promote binding of distinct structural forms of VP40. For instance, VP40 forms an octameric ring structure that regulates viral transcription12 and lipid binding of the ring structure has not been explored. PIP interactions with the octameric ring could also be an important part of the viral life cycle as VP40 has been shown to enter the nucleus59 where several PIPs reside. Finally, lipids may also play a role in VP40 trafficking to the plasma membrane60. Currently, the role of PS or other PIP species in intracellular VP40 transport is unknown. In the experiments presented here and previously24 a decrease in available PS yields a decrease in VLP egress and localization (Fig. 2); however, PS localization is not limited to the plasma membrane. Therefore, PS on secretory vesicles from the Golgi or endocytic recycling vesicles could also have an important role in VP40 trafficking to the plasma membrane. Additionally, while VP40 does not bind to MLVs containing PI4P (Fig. 5C,D), excess SidM-P4M blocks VP40 PM localization. It is still possible that PI(4)P has an indirect role in VP40 trafficking. In contrast, VP40 does bind the monophosphate PI(3)P (Fig. 5C,D), which could have an important role in the life cycle of the virus or VP40 function, independent of plasma membrane assembly and budding as PI(3)P is abundant on early endosomes28 and important for autophagy.
There is still much to learn in how EBOV and VP40 interacts with host cell lipids and lipid synthesizing machinery. A greater understanding of these interactions should provide greater insight into the function of lipids in VP40 trafficking, plasma membrane localization, oligomerization, viral particle assembly and viral egress. Resolving the molecular basis of lipid head group and membrane bilayer association of VP40 with high-resolution structural techniques could provide a more complete picture of the mechanisms by which VP40 structures assemble and rearrange at the plasma membrane inner leaflet.
Materials and Methods
Plasmids
The VP40-pET46 expression vector was a kind gift from Erica Ollmann Saphire (The Scripps Research Institute) and EGFP-VP40 was prepared as previously described21. The Lact-C2 EGFP, Lact-C2 mCherry, KRΦ-mRFP and Rpre-mRFP plasmids were kind gifts from Dr. Sergio Grinstein (University of Toronto). PLC□ PH GFP was a gift from Tamas Balla (NIH) while the lyn11-FRB-CFP, PJ-WT-FKBP, PJ-INPP5E-FKBP, PJ-Sac1-FKBP and PJ-Dead-FKBP and SidM-P4m-GFP, SidM-P4Mx2-GFP, SidM-P4M-mCherry were kind gifts from Tamas Balla (NIH) and Gerald Hammond (University Of Pittsburgh). Akt1 PH GFP was from Tobias Meyer (Stanford University), HIV-1 GAG EGFP was a kind gift from Tom Hope (Northwestern University). All plasmids were transformed into DH5α cells and purified using an MIDI-Prep Kit (QIAGEN (Valencia, CA)) with endotoxin free buffers (QIAGEN).
Cell Culture and Transfection
COS-7 cells were used for all experiments. They were maintained at 37 °C in a 5% CO2 humidified incubator supplemented with DMEM with 1 g/L L-Glutamine, D-Glucose, 110 mg/mL Sodium Pyruvate (Life Technologies (Carlsbad, CA)) containing 10% FBS (Sigma (St. Louis, MO)) and 1% Penicillin-Streptomycin (Life Technologies). In preparation for imaging experiments cells were washed with 10 mL 1X PBS, pH 7.4 and trypsinized with 1X Trypsin-EDTA (MP Biomedicals (Santa Ana, CA)). Cells were then seeded in No. 1.5 glass bottom 8 well plates (MatTek (Ashland, MA) and grown to 80–90% confluency before transfection. Cells were transfected with endotoxin free plasmid DNA using Lipofectamine 2000 (Life Technologies) in optiMEM (Life Technologies) according to manufacturers protocol for 14 hours at 37 °C unless indicated otherwise. 0.4ug of DNA was used of each plasmid per transfection for pharmaceutical or lipid treatment. In experiments with one or more plasmid per cell 0.4ug DNA was used for the construct being tested, 0.15 ug DNA in “low” transfection for the competition assay and 0.5 ug for the “excess” transfection in the competition assay. 0.5 ug DNA were used for the enzymatic constructs used in combination with VP40 and other proteins (0.4 ug DNA). For VLP collection experiments and SEM experiments the amount of DNA and Lipofectamine 2000 were scaled up proportionally with sample size.
Confocal Microscopy
Imaging experiments were performed with a Zeiss LSM 710 inverted microscope using a Plan Apochromat 63× 1.4 numerical aperture oil objective. A 458 nm argon laser was used to excite CFP, 488 nm argon laser was used to excite GFP/EGFP and a 561 nm diode laser was used to excite RFP and mCherry. Laser power was kept constant for all experiments. Three independent experiments were performed for all experiments unless indicated otherwise.
Image Analysis
ImageJ or MATLAB (MathWorks (Natick, MA)) were used for all data analysis. A custom MATLAB script (available upon request) was used to determine the average intensity at the PM and cytosol of each cell. In the script the average intensity in the plasma membrane (defined by the outer 6 pixels of each cell) and the cytosol (region within the PM region) were calculated. The values for PM and cytosol intensity for each cell were then used to calculate the %PM localization. The %PM localization was calculated with the following equation: PM intensity over the total (PM+Cytosol) intensity ×100%. Values were normalized to controls (control set to 100%) and the standard error of the mean was scaled accordingly. Because this script uses the outer 6 pixels of each cell, a value of 0% PM localization cannot be achieved. When cytosolic EGFP is transfected into COS7 cells, there is a value of 14.8 ± 1.2% PM localization, N = 10. For EGFP-VP40, the %PM localization before normalizing to 100% was 36.3 ± 2.3%, N = 22. When compared to cytosolic EGFP, if EGFP-VP40 has a %PM localization of 14.8 ± 1.2%, there is no true localization. When the data is normalized to 100%, EGFP-VP40 value of 40% PM localization or lower is equivalent to cytosolic EGFP when EGFP is normalized to VP40 (14.8/36.3 = 40.7). A value below 40% (normalized) is usually observed in cells with high intensity structures in the cytosol or internal membranes of the cell. The standard error of the mean was calculated for all datasets and scaled proportionally to the normalized data (SEM*(Normalized Value/Original Value) = Scaled SEM). Data sets are shown ± scaled SEM. A two tailed students t-test was used to determine significance (p < 0.05).
VLP Collection
COS-7 cells were grown to 90 percent confluency in 100 mm plates and transfected with Lipofectamine 2000 (Life Technologies) and endotoxin free plasmid DNA in optiMEM (Life Technologies) for 24 hours. Media from the transfected plates were removed to harvest VLPs plates were then washed with 10 mL 1x PBS, pH 7.4. VLP Samples (media + 1×PBS wash) were centrifuged for 6 minutes at 4 °C at 900 rpm (Eppendorf 5810 R 15 Amp version) to remove dead cell debris from the sample. Supernatant was then gently pipetted on top of a 20% sucrose cushion and samples were then centrifuged for 120 minutes at 100,000 × g, 4 °C (Beckman Coulter Avanti J-30I centrifuge) to pellet VLPs. VLPs were dried and resuspended in 150 mM ammonia bicarbonate (NH4HCO3) and stored at −80° C. During the 120 minute VLP centrifugation step, cells were trypsinized off of the plates and pelleted for 6 minutes at 4 °C at 900 rpm. Pellets were washed with 1x PBS and cells were counted (BioRad TC10 Automated Cell Counter). Cells were then pelleted again and supernatant was removed. Pellets were then resuspended in a COS-7 specific lysis buffer adapted from Thermo Scientific (50 mM TRIS-HCl, 150 mM NaCl, 1 mM EDTA, 1% TritonX, 0.1% SDS, 2 uM PMSF, 1% Deoxycholic acid and 1% 100 × protease inhibitor cocktail). Cells were incubated on ice in the lysis buffer for 60 minutes with vortexing every 15 minutes. Cell lysate (CL) samples were centrifuged at 25,000 × g for 17 minutes at 4 °C (Beckman Coulter Avanti J-30I centrifuge). The soluble fraction was removed and stored at −80 °C.
GFP ELISA
A GFP ELISA assay (Cell Biolabs (San Diego, CA)) was used to quantify the GFP in the VLP and cell lysate (CL) samples from VLP collections. The GFP ELISA was performed according to manufactures instructions. ELISA plates were analyzed by measuring absorbance of each microwell on a spectrophotometer (Molecular Devices SpectraMax M5) using 450 nm as the primary wave length. Protein content of VLP and CL samples were determined using a BCA assay (Pierce (Grand Island, NY)) to assist in sample VLP and CL sample dilution. All samples were diluted in assay diluent between 40 and 5000 times to get sample between 10–2000 pg GFP (linear range of this GFP ELISA kit). All standards and samples were performed in duplicate. The %VLP egress was quantified by determining the GFP concentration in the VLP sample over the total GFP (VLP+CL) *100% for each sample. Data represents at least three independent transfections with VLP collections on at least two different days.
Scanning Electron Microscopy (SEM)
Samples were prepared for SEM by transfecting COS-7 cells using the transfection procedure described in cell culture and transfection. 14 hours post transfection cells were scraped from the tissue culture dish, pelleted and washed with 1× PBS. After pelleting again, cells were resuspended in primary fixative (2% glutaraldehyde, 2% paraformaldehyde in 0.1 M cacodylate buffer pH 7.35) and incubated for at least 14 hours at 4 °C on an orbital rocker. Samples were then pelleted and resuspended in 0.1 M cacodylate buffer and samples were applied to poly-L-lysine coated 13 mm coverslips. 1% osmium tetroxide was applied to samples, followed by water washes and dehydration using ethanol. Samples were they dried using a Tousimis Autosamdri-931 (Rockville, MD). Before imaging, 3 nm iridium was applied to the samples, images were collected on a Field Emission Scanning Electron Microscope Magellan 400 (FEI) (Hillsboro, OR) with assistance from Tatyana Orlova and the Notre Dame Integrated Imaging Facility.
Pharmacological and Lipid Treatments
Plasma membrane neutralization: D-erythro-Sphingosine (Avanti Polar Lipids Inc.) was dried under N2 and resuspended in ethanol. Cells were treated with 75 uM sphingosine or ethanol vehicle (1:250 vol/vol) for 1 or 12 hours at 37 °C. Data were normalized to ethanol vehicle. Ionomycin treatment: cells were treated with 10 uM ionomycin (Calbiochem (San Diego, CA)) in DMSO or DMSO vehicle were added to cells in glucose free media with 2 mM CaCl2 or 1× Annexin V binding buffer (BD Biosciences (Franklin Lakes, NJ)) for five minutes. Cells were imaged pre-treatment and each minute post treatment for a timely quantitative analysis. Annexin V-APC (BD Biosciences) 1:40 was added five minutes post treatment and imaged to confirm PS exposure. Phenylarsine oxide (PAO): Cells were treated with 10 uM PAO (Sigma) in DMSO or DMSO vehicle were added to transfected cells for 30 minutes. Data was normalized to vehicle treatment. Cells were treated with 100 nM Wortmannin (Sigma) or equivalent volume DMSO for one hour at 37 °C.
Rapamycin Induced Lipid Depletion System
Transfected cells with Lyn11-FRB-CFP, PJ-(WT, INPP5E, Sac1 or Dead)-FKBP-mRFP and either PLCδ-PH-EGFP, LactC2-EGFP, EGFP-VP40 or HIV-GAG-EGFP were treated with 1 uM (final concentration) rapamycin (Sigma) for 7 minutes during imaging on a Zeiss LSM 710 as described above. Data analysis was performed in MATLAB as described above. The %PM localization was determine pre and post rapamycin addition.
Protein Purification
Ebola WT-VP40 in the pET46 Ek/LIC (His-tagged) (Novagen) vector was expressed and purified as previously described by Bornholdt et al.12. Bacterial pellets were lysed with a combination of lysozyme and sonication. The protein was purified by binding with Ni/NTA resin slurry and centrifugation at 400 × g. Purified protein was dialyzed into (1.5 M KCl, 250 mM HEPES, pH 7.4) buffer and concentrated with a 10,000 MWCO filter. A sample of the pure protein was run on an SDS-PAGE gel and Coomassie stained to confirm appearance of a strong band at 40 kDa. Concentration was calculated with a BCA Assay and the concentrated protein stored at 4 °C for a maximum of 14 days.
Multilamellar Vesicle (MLV) Sedimentation Assay
MLV emulsion was prepared by combining phosphatidylcholine (POPC), phosphatidylethanolamine (POPE), phosphatidylserine (POPS) and PIPs in the specified proportions and drying to a film under nitrogen. The film was then hydrated with VP40 dialysis buffer (150 mM KCl, 25 mM HEPES, 0.1 mM EGTA, pH 7.4) to a lipid concentration of 1.0 mM and incubated for 10 minutes at 37 °C to expedite lipid hydration. The hydrated lipids were vortexed for 60 seconds to form a suspension of MLVs. Suspension and protein were combined to a final protein concentration of 5uM and final volume of 100uL per sample. The mixture was incubated at room temperature for 20 minutes. Following centrifugation at 75,600 × g for 20 minutes, the supernatant (soluble protein) was collected. The pellet (lipid associated protein) was resuspended in VP40 dialysis buffer to the same volume as the supernatant and vortexed for 15 seconds. 40 uL of each sample was loaded onto an SDS-PAGE gel and run for 60 minutes at 150 V, 400 mA. Gels were stained with Coomassie Brilliant Blue, imaged and analyzed with ImageJ software to determine percentage of lipid associated protein. The experiment was performed with four replicates per MLV composition and experimental triplicate (n = 12).
Raster Image Correlation Spectroscopy (RICS)
Images acquired for RICS analysis were collected on an Olympus FluoView FV1000 microscope with a 40 × 1.3 numerical aperture oil objective. Raster scanning with 12.5 μs pixel dwell and 16.4 zoom (for 50 nm pixel size) was used to collect 100 images of each cell according to the methods described by Rossow et al.61. Data analysis was performed in SimFCS software according to the protocol of Rossow et al.61. GFP in solution was used to calibrate the focal volume of the laser beam waist (Wo) each day data was collected. The diffusion of GFP in solution is known61 and held constant to determine the Wo value between 0.2 and 0.4. Once Wo is determined, it is held constant and diffusion coefficients are determined for each construct. The diffusion coefficients are averaged over three independent experiments and the standard error of the mean was calculated for each and significance determined using a two-tailed t-test.
Number and Brightness Analysis
Images acquired for N&B analysis were collected as described for RICS. Data Analysis was performed in SimFCS software using the number and brightness analysis tool. This method allows for the discrimination between intensity of a pixel and the brightness of a pixel by measuring the variance of intensity at each pixel over time. This method determines the oligomerization status (brightness) versus intensity of each pixel within the image that can be used to determine various oligomer states of a protein throughout a cell46. True brightness of a monomer in COS7 cells was determined using monomeric EGFP (N = 11). A value of 0.122 ± 0.017 was calculated from the peak of the Gaussian brightness distribution plot in SimFCS when the S factor was set to 2.5. Equation 1 was used to calculate aggregate62.

The number of pixels of monomer-hexamer, hexamer-12mer and 12mer+ were calculated for each condition and normalized to the control, EGFP-VP40. The standard error of the mean (SEM) was calculated for each and significance was determined using a two-tailed t-test. Average values are shown ± SEM.
Additional Information
How to cite this article: Johnson, K. A. et al. The Ebola Virus matrix protein, VP40, requires phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci. Rep. 6, 19125; doi: 10.1038/srep19125 (2016).
References
Feldmann, H. & Geisbert, T. W. Ebola haemorrhagic fever. Lancet 377, 849–862 (2011).
Jasenosky, L. D., Neumann, G., Lukashevich, I. & Kawaoka, Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol 75, 5205–5214 (2001).
Noda, T. et al. Ebola Virus VP40 Drives the Formation of Virus-Like Filamentous Particles Along with GP. J. Virol 76, 4855–4865 (2002).
Harty, R. N. No exit: targeting the budding process to inhibit filovirus replication. Antiviral research 81, 189–197 (2009).
Shtahelin, R. V. Could the Ebola virus matrix protein VP40 be a drug target? Expert Opin Ther Targets 18, 215–220 (2014).
Spurgers, K. B. et al. Identification of Essential Filovirion-associated Host Factors by Serial Proteomic Analysis and RNAi Screen. MCP 9 (2010).
Yamayoshi, S. et al. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell host microbe 3, 168–177 (2008).
Han, Z. et al. ALIX Rescues Budding of a Double PTAP/PPEY L-Domain Deletion Mutant of Ebola VP40: A Role for ALIX in Ebola Virus Egress. J. Infect. Dis (2015).
Timmins, J. et al. Ebola Virus Matrix Protein VP40 Interaction with Human Cellular Factors Tsg101 and Nedd4. JMB 326, 493–502 (2003).
Ruthel, G. et al. Association of ebola virus matrix protein VP40 with microtubules. J. Virol 79, 4709–4719 (2005).
Harty, R. N., Brown, M. E., Wang, G., Huibregtse, J. & Hayes, F. P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. USA 97, 13871–13876 (2000).
Bornholdt, Z. A. et al. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell 154, 763–774 (2013).
Dessen, A., Volchkov, V., Dolnik, O., Klenk, H.-D. & Weissenhorn, W. Crystal Structure of the matrix protein VP40 from Ebola Virus. EMBO J 19, 4228–4236 (2000).
Radzimanowski, J., Effantin, G. & Weissenhorn, W. Conformational plasticity of the Ebola virus matrix protein. Protein Sci 23, 1519–1527 (2014).
Gomis-Rüth, F. X. et al. The Matrix Protein VP40 from Ebola Virus Octamerizes into Pore-like Structures with Specific RNA Binding Properties. Structure 11, 423–433 (2003).
Hoenen, T. et al. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol 84, 7053–7063 (2010).
Soni, S. P., Adu-Gyamfi, E., Yong, S. S., Jee, C. S. & Stahelin, R. V. The Ebola virus matrix protein deeply penetrates the plasma membrane: an important step in viral egress. Biophy. J 104, 1940–1949 (2013).
Adu-Gyamfi, E. et al. The Ebola virus matrix protein penetrates into the plasma membrane: a key step in viral protein 40 (VP40) oligomerization and viral egress. J. Biol. Chem 288, 5779–5789 (2013).
Adu-Gyamfi, E. et al. A loop region in the N-terminal domain of Ebola virus VP40 is important in viral assembly, budding and egress. Viruses 6, 3837–3854 (2014).
Silva, L. P., Vanzile, M., Bavari, S., Aman, J. M. & Schriemer, D. C. Assembly of Ebola virus matrix protein VP40 is regulated by latch-like properties of N and C terminal tails. PloS one 7, e39978 (2012).
Adu-Gyamfi, E., Digman, M. A., Gratton, E. & Stahelin, R. V. Investigation of Ebola VP40 assembly and oligomerization in live cells using number and brightness analysis. Biophys J 102, 2517–2525 (2012).
Ruigrok, R. W. et al. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J Mol Biol 300, 103–112 (2000).
Scianimanico, S., Schoehn, G., Ruigrok, R. & Weissenhorn, W. Membrane association induces a conformational change in the Ebola virus matrix protein. EMBO J 19, 6732–6741 (2000).
Adu-Gyamfi, E. et al. Host Cell Plasma Membrane Phosphatidylserine Regulates the Assembly and Budding of the Ebola Virus. J. Virol (2015).
Kondratowicz, A. S. et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 108, 8426–8431 (2011).
Moller-Tank, S. & Maury, W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology 468–470, 565–580 (2014).
Morizono, K. & Chen, I. S. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol 88, 4275–4290 (2014).
Hammond, G. R. & Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758 (2015).
Xu, C., Watras, J. & Loew, L. M. Kinetic analysis of receptor-activated phosphoinositide turnover. J. Cell. Bio 161, 779–791 (2003).
Willars, G. B., N. S. R. & John, R. A. Challis Differential Regulation of Muscarinic Acetylcholine Receptor- sensitive Polyphosphoinositide Pools and Consequences for Signaling in Human Neuroblastoma Cells. J. Biol. Chem 273, 5037–5046 (1997).
Balla, T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev 93, 1019–1137 (2013).
Vance, J. E. Phospholipid synthesis and transport in mammalian cells. Traffic 16, 1–18 (2015).
Barlow, C. A., Laishram, R. S. & Anderson, R. A. Nuclear phosphoinositides: a signaling enigma wrapped in a compartmental conundrum. Trends in cell biol 20, 25–35 (2010).
Watt, S. A., K. G., Fleming, I. N., Downes, C. P. & Lucocq, J. M. Subcellular localization of phosphatidylinositol 4,5-bisphosphate using the pleckstrin homology domain of phospholipase C δ1. Biochem. J. 363 (Pt 3), 657–666 (2002).
Irvine, R. F. Nuclear lipid signalling. Nat. Rev. Mol. Cell Biol. 4, 349–360 (2003).
Chukkapalli, V., Hogue, I. B., Boyko, V., Hu, W. S. & Ono, A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient gag membrane binding. J. Virol 82, 2405–2417 (2008).
Saad, J. S. et al. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J Mol Biol 382, 434–447 (2008).
Prchal, J., Kroupa, T., Ruml, T. & Hrabal, R. Interaction of Mason-Pfizer monkey virus matrix protein with plasma membrane. Front. Microbiol 4, 423 (2013).
Stansell, E. et al. Basic residues in the Mason-Pfizer monkey virus gag matrix domain regulate intracellular trafficking and capsid-membrane interactions. J. Virol 81, 8977–8988 (2007).
Stauffer, T. P., Ahn, S. & Meyer, T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol 8, 343–346 (1998).
Tony Yeung, et al. Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science 319, 210–213 (2008).
Heit, B., Yeung, T. & Grinstein, S. Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis. Am J Physiol Cell Physiol 300, C33–C41 (2011).
Brunner, J. D. et al. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 (2014).
Hammond, G. R. et al. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 337, 727–730 (2012).
Toker, A. & Cantley, L. C. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387, 673–676 (1997).
Digman, M. A., Dalal, R., Horwitz, A. F. & Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J 94, 2320–2332 (2008).
Stahelin, R. V. Could the Ebola virus matrix protein VP40 be a drug target? Expert Opin. Ther. Targets 18, 115–120 (2014).
Panchal, R. G. et al. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. Proc. Natl. Acad. Sci. USA 100, 15936–15941 (2003).
Hamard-Peron, E. et al. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J. Virol 84, 503–515 (2010).
Vanhaesebroeck et al. Synthesis and Function of 3-Phosphorylated Inositol Lipids. Annu Rev Biochem 70, 535–602 (2001).
Alessandra Borsetti, A. S. O. & Heinrich, G. Gottlinger The C-Terminal Half of the Human Immunodeficiency Virus Type 1 Gag Precursor Is Sufficient for Efficient Particle Assembly. J. Virol 72, 9313–9317 (1998).
Ono, A., Ablan, S. D., Lockett, S. J., Nagashima, K. & Freed, E. O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 101, 14889–14894 (2004).
Balaji Olety, A. O. Roles played by acidic lipids in HIV-1 Gag membrane binding. Virus Res 193 (2014).
Dick, R. A., Kamynina, E. & Vogt, V. M. Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J. Virol 87, 13598–13608 (2013).
Chukkapalli, V., Inlora, J., Todd, G. C. & Ono, A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J. Virol 87, 7155–7159 (2013).
Chukkapalli, V., Oh, S. J. & Ono, A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. USA 107, 1600–1605 (2010).
Vance, J. E. & Steenbergen, R. Metabolism and functions of phosphatidylserine. Prog. Lipid Res 44, 207–234 (2005).
Kay, J. G. & Grinstein, S. Sensing phosphatidylserine in cellular membranes. Sensors 11, 1744–1755 (2011).
Nanbo, A., Watanabe, S., Halfmann, P. & Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep 3, 1206 (2013).
Kolesnikova, L., Bamberg, S., Berghofer, B. & Becker, S. The Matrix Protein of Marburg Virus Is Transported to the Plasma Membrane along Cellular Membranes: Exploiting the Retrograde Late Endosomal Pathway. J. Virol 78, 2382–2393 (2004).
Rossow, M. J., Sasaki, J. M., Digman, M. A. & Gratton, E. Raster image correlation spectroscopy in live cells. Nat. Protoc 5, 1761–1774 (2010).
Rose, E., Dixon, C. Y., Edward, P., Cheng, Manuel, F. N. & Luis, F. S. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl. Acad. Sci. USA 109, 1749–1754 (2011).
Acknowledgements
This work was partially supported by the NIH (AI081077) to R.V.S. R.V.S. also acknowledges support from the Indiana University School of Medicine-South Bend Imaging and Flow Cytometry Core facility. J.L.S. was supported by an AHA predoctoral fellowship (12PRE11960030) and a NIH CBBI predoctoral fellowship (T32GM075762). COS-7 cells were a kind gift from Dr. Holly Goodson (Notre Dame). K.A.J thanks Sergio Grinstein, Gerald Hammond and Michelle Digman for helpful discussions. K.A.J. thanks Yuliya Klymenko and Sharon Stack for their expertise in SEM sample prep and generous sharing of SEM prep methods and reagent. K.A.J thanks Tatyana Orlova and the Notre Dame Integrated Imaging Facility for her assistance and excellence in SEM image acquisition.
Author information
Authors and Affiliations
Contributions
K.A.J. and R.V.S. conceived of the studies and designed all experiments. K.A.J. and G.T. performed experiments and data analysis. K.A.J. and R.V.S. wrote the manuscript with input from all authors. J.L.S. gave valuable experimental insight.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Johnson, K., Taghon, G., Scott, J. et al. The Ebola Virus matrix protein, VP40, requires phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) for extensive oligomerization at the plasma membrane and viral egress. Sci Rep 6, 19125 (2016). https://doi.org/10.1038/srep19125
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19125
This article is cited by
-
Altered Lipid Metabolism in Recovered SARS Patients Twelve Years after Infection
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
