
Abstract
The cystathionine β-synthase (CBS) gene has been shown to be related to homocystinuria. This study was aimed to detect the mutations in CBS in a Han Chinese family with homocystinuria. A four-generation family from Shandong Province of China was recruited in this study. All available members of the family underwent comprehensive medical examinations. Genomic DNA was collected from peripheral blood of all the participants. The coding sequence of CBS was amplified by polymerase chain reaction (PCR), followed by direct DNA sequencing. Among all the family members, three affected individuals showed typical clinical features of homocystinuria. Two novel compound heterozygous mutations in the CBS gene, c.407T > C (p. L136P) and c.473C > T (p.A158V), were identified by sequencing analysis in this family. Both of the two missense mutations were detected in the three patients. Other available normal individuals, including the patients’ parents, grand parents, her younger sister and brother in this family either carried one of the two mutations, or none. In addition, the two mutations were not found in 600 ethnically matched normal controls. This study provides a mutation spectrum of CBS resulting in homocystinuriain a Chinese population, which may shed light on the molecular pathogenesis and clinical diagnosis of CBS-associated homocystinuria.
Similar content being viewed by others


Introduction
Homocystinuria, most commonly caused by cystathionine β-synthase (CBS) deficiency, is an autosomal recessive disorder of sulfur amino acid metabolism. CBS protein is a pyridoxal 5′ phosphate dependent enzyme and catalyzes the condensation of homocysteine with homocysteine and serine to form cystathionine1. Biochemically, this disorder is characterized by elevated plasma concentrations of homocysteine and methionine, increased excretion of homocysteine in urine and decreased levels of cystathionine and cysteine in body fluids2. Patients with homocystinuria often display different symptoms,including ocular anomalies (severe myopia and ectopialentis), skeletal deformities (osteoporosis, scoliosis and Marfanoid habitus), vascular thrombosis and ischemia, disorder of central nervous system (mental retardation, convulsions and psychiatric disturbances) and other manifestations3.
It has been reported that homocystinuria due to CBS deficiency is caused by mutations in the CBS gene4. CBS mutations could lead to the disruption of enzyme activity which consequently results in increased levels of homocysteine, a potentially toxic amino acid responsible for patients with homocystinuria. So far, more than 150 mutations in the CBS gene have been identified (http://cbs.lf1.cuni.cz/mutations.php) in different ethnic population5,6,7,8,9,10,11,12. Although many mutations have been described in the CBS gene from homocystinuric patients and this disease has been well characterized in other populations, the range of clinical presentations and spectrum of CBS mutations in Han Chinese patients remained largely uninvestigated.
In this study, we characterized the clinical manifestations and investigated the molecular basis of a Han Chinese family with homocystinuria, to expand the CBS mutation spectrum of the homocystinuric patients from China.
Materials and Methods
Subjects
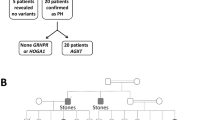
This family with homocystinuria, including 16 members, was recruited from Shandong Provincial Hospital Affiliated to Shandong University (Fig. 1). Three of the family members were diagnosed as homocystinuria by biochemical profiles (grossly increased serum homocysteine and methionine) and by critical complications, such as dislocated lens, mental retardation and skeletal deformities. Their clinical information is summarized in Table 1.This study was conducted in accordance with the tenets of the Declaration of Helsinki and approved by the Institutional Review Boards of Hospital of University of Electronic Science and Technology of China & Sichuan Provincial People’s Hospital and Shandong Provincial Hospital Affiliated to Shandong University. Written informed consents were obtained from the family prior to the study.Unrelated healthy control subjects were recruited from the Hospital of University of Electronic Science and Technology of China and Sichuan Provincial People’s Hospital. These controls are all Han Chinese and subjects were excluded from the study if they had any of the symptoms, including ocular anomalies, skeletal deformities, vascular thrombosis and disorder of central nervous system.

Pedigree of the family with homocystinuria.
Solid symbols indicated affected individuals and open symbols indicate unaffected individuals. Arrow indicates the proband.
DNA Extraction
All genomic DNA was extracted from peripheral blood using a blood DNA extraction kit (QIAamp DNA Blood Midi Kit; Qiagen, Germany) according to the manufacturer’s protocol. DNA samples were stored at −20 °C until used. DNA integrity was evaluated by 1% agarose gel electrophoresis.
Mutation screening
The method for mutation screening was performed as described previously13. Besides the variants in CBS, mutations in the MTHFR gene also have been reported to cause homocystinuria14,15. Therefore, the coding sequences of CBS (NM_000071.2) and MTHFR(NM_005957.4) were amplified by polymerase chain reaction (PCR) using a MyCycler thermo cycler (Bio-Rad, Hercules, CA). We did not detect any mutation in the MTHFR gene in this family (data not shown), thus it was excluded to cause homocystinuria in this study. Sequencing primers from flanking sequence of each exon of the CBS gene were designed by using the Primer 5.0 (Table 2). Amplification reaction was performed by the PCR reaction (10 μL final volume) containing 50 ng of genomic DNA, 1 μL of each primer (10 pmol/μL), 1 μL of 10 buffer (Takara Bio Inc., Shiga, Japan), 0.8 μL of deoxyribonucleotide triphosphates (2 mmol/L; Takara Bio Inc.), 0.4 μL MgCl2 (2.5 mmol/L; Takara Bio Inc.) and 0.1 μL of ExTaq polymerase (5 U/μL; Takara Bio Inc.). Amplified PCR products were purified with spin columns (QIAquick, Qiagen, Valencia, CA) and sequenced directly (BigDye Terminators Sequencing Kit; Applied Biosytems) in both directions with an automated genetic analysis system (ABI 3130 Genetic Analyzer, CA, USA).
Multiple sequence alignment of the human CBS protein was performed along with other CBS protein across different species, to check for the conservation of the residues. The possible damaging effects of the 2 mutations on the structure and function of CBS were predicted using SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
Results
Clinical findings
A four-generation family from Shandong Province of China was recruited in this study (Fig. 1). There are three affected individuals (III:2, III:3 and III:4), who showed typical clinical symptoms of homocystinuria among all the family members. The proband (III:3), as well as her two affected sisters(III:2 and III:4) exhibited similar clinical features, such as various reduced visual acuities with a bilateral lens dislocation, myopia, glaucoma, skeletal deformities and mental retardation(Table 1). All the patients also have elevated plasma homocysteine and methionine levels, compared to all the normal individuals of this family (±13.6 μmol/L for homocysteine and ±24.7 μmol/L for methionine, respectively). The parents, the grand parents and other relatives of the three affected individuals had no homocystinuric symptoms, exhibiting a pattern of recessive inheritance in this family.
Mutation screening of CBS in homocystinuria
Sequencing analysis of the CBS gene revealed novel compound heterozygous mutations, c.407T > C(p. L136P) and c.473C > T (p.A158V) (Fig. 2). They located in the coding sequence at nucleotide 407 of exon 3 and at nucleotide 473 of exon 4, respectively (Fig. 3). Both the two missense mutations were present in the three affected subjects (Table 3). The parents of the three patients were unaffected carriers with c.473C > T(father) and c.407T > C(mother) mutations, showing complete co-segregation of the mutations with the disease phenotype. Other available normal individuals in this family either carried one of the two mutations, or none. In addition, neither of the two missense heterozygous mutations was detected in 600 ethnically matched normal controls.

Direct sequencing results of CBS mutations.
Direct sequencing identified two novel compound heterozygous mutations, (A) c.407T > C(p. L136P) and (B) c.473C > T (p.A158V)(indicated by red arrow).

Mutations in the CBS gene identified in Chinese homocystinuric patients.
The boxed mutations in red were newly found in this study and the mutations in blue box were identified in a Hong Kong homocystinuric patient.
Comparative amino acid sequence alignment of other CBS protein across different species revealed that the two novel mutations occurred at highly conserved positions (Fig. 4). Both of the two novel mutations could result in substitutions of amino acid in the CBS protein and were predicted to be damaging by SIFT and Polyohen 2 (Table 3). The c.407T > C mutation is a T-C transition, converting Leucine (L) to Proline (P) at amino acid 136 (p. L136P); another mutation is a C-T transition (c.407T > C), leading to substitution of Alanine (A) toValine (V) at codon 158 (p.A158V, Fig. 2).

Orthologous protein sequence alignment of CBS from different species.
The mutated residue showing conservation was shaded in red. Red shaded amino acids proteins showed that the two novel missense mutations occurred at highly conserved positions in these species.
Discussion
Homocystinuria is the most common inborn disorder of sulfur amino acid metabolism. CBS deficiency, a main factor causing homocystinuria, is an autosomal recessively inherited genetic defect. Since the first mutation in the human CBS gene reported by Kozich and Kraus in 19924, many CBS mutations in homocystinuric patients from various populations worldwide have been identified. The present study identified novel compound heterozygous mutations, c.407T > C (p. L136P) and c.473C > T (p.A158V), in a Han Chinese family with homocystinuria and this result expands the spectrum of CBS mutations resulting in homocystinuria.
The human CBS gene, located at chromosome 21q22.316, consists of 63-kDa subunits and encodes an enzyme with 551 amino acids17. The enzyme’s structure consists of a catalytic domain with 409 amino acids in the N-terminal and a regulatory domain with 142 amino acids in the C-terminal18. The protein encoded by this gene acts as a homotetramer to catalyze the conversion of homocysteine to cystathionine, the first step in the transsulfuration pathway1. The encoded protein is allosterically activated by adenosyl-methionine and uses pyridoxal phosphate as a cofactor. Defects in this gene can cause CBS deficiency, which can lead to homocystinuria. And most of affected patients are compound heterozygotes of these CBS mutations10,14,15,19. Until now, molecular genetic analyses of CBS deficiency have identified more than 150 pathogenic mutations among homocystinuric patients, mostly in the Caucasian populations and very few in African-Americans and Asians20. In 2011, two CBS mutations (c.833T > C and c.1006C > T) were detected in a Hong Kong homocystinuric patient by Kwok et al.19, however, it is so far the only report describing mutations in the CBS gene in Chinese (Fig. 2). In addition to the mutations identified in this study, the spectrum of mutations in CBS observed Han Chinese bears less resemblance to those observed in in Japanese and Korean patients7,9.
In this study, mutation analysis of three patients with homocystinuria in a Han Chinese family is described and we identified novel compound heterozygous for mutations c.407T > C (p. L136P) in exon 3and c.473C > T (p.A158V) in exon 4 of the CBS gene (Fig. 3). So far, these two mutations are reported in homocystinuric patients for the first time in mainland Han Chinese, although CBS mutations have been identified in different ethnic groups. In this pedigree, all the three affected patients (III:2, III:3 and III:4) were found to harbor both of the two missense mutations in CBS. The patients in this family were diagnosed as homocystinuria based on detection of elevated blood homocysteine and diagnosis of skeletal deformities, mental retardation and ectopialentis. The proband of this family (III:3) presented with reduced vision and were diagnosed by Provincial Hospital Affiliated to Shandong University at the age of 15. Biocularlens dislocation, high myopia, exotropia, glaucoma and corneal staphyloma were proved at that time. She had a history of bilateral downward dislocation of the lens since 7years old. No significant family history was noted except her two sisters (III:2 and III:4), who exhibited similar clinical manifestations with the proband. Surgeries were performed to extract the dislocated lens during 8 to 11 years old in these three affected girls. Their plasma total homocysteine level and methionine level were both markedly elevated, confirming the diagnosis of homocystinuria (Table 3). Molecular genetic testing of the CBS gene also helps to confirm the diagnosis of patients. Mutation analysis was also performed on the patient’s parents as well as her younger sister and brother, who are all unaffected (Fig. 1). Sequencing analysis showed that the father (II:3) and her sister (III:5)only carried thec.473C > T (p.A158V) mutation. Her mother (II:4) and brother(III:6) were heterozygous for thec.407T > C (p. L136P) mutation (Table 3).In addition, none of the two mutations in CBS was detected in 600 normal controls through gene analysis. Considering that CBS deficiency is an autosomal recessive disorder and that no other alteration was detected in coding regions of the CBS gene in homocystinuric patients of this family, it is highly possible that the two novel mutations of CBS identified here are responsible for the pathogenesis of homocystinuria in this pedigree.
For the p.L136P mutation identified in this pedigree, Leucine was replaced by Proline in exon 3, which is the most evolutionary conserved part of the CBS enzyme. Among all mutations identified in homocystinuric patients, about 25% mutations were in this conserved region (the third exon of this gene)5,6,7,8,9,10,11,12. The p.A158V mutation of CBS resulted in a substitution of Alanine to Valinein exon 4. Moreover, they are both predicted to be probably damaging to protein function by SIFT and PolyPhen-2. However, the exact mechanisms and pathological roles of the two novel mutations in CBS in the development of homocystinuria are largely unknown. In addition, the spectrum of mutations observed in this study bears less resemblance to those observed in Japanese9, Korean7 and Filipino10 patients, as well as Western countries, suggesting possible existence of ethnic differences among various populations. Future studies on Chinese homocystinuric subjects may help to provide more evidence for this hypothesis. In order to better understand homocystinuria pathogenesis, functional studies are needed to illustrate the role of CBS and the underlying mechanisms of this disease.
These data, together with the clinical presentation of the three affected siblings, demonstrated that p.L136Pandp.A158Vmutations in the CBS gene were responsible for homocystinuria in a Han Chinese family. Our data of CBS mutations causing homocystinuria further confirm the role of CBS in the pathogenesis of homocystinuria. This study expands the mutation spectrum of CBS resulting in homocystinuria, which could provide insights into the pre-symptomatic molecular diagnosis, the management of homocystinuric patients and the genetic counseling of families in Chinese.
Additional Information
How to cite this article: Gong, B. et al. Novel Compound Heterozygous CBS Mutations Cause Homocystinuria in a Han Chinese Family. Sci. Rep. 5, 17947; doi: 10.1038/srep17947 (2015).
References
Miles, E. W. & Kraus, J. P. Cystathionine beta-synthase: structure, function, regulation and location of homocystinuria-causing mutations. J Biol Chem 279, 29871–29874 (2004).
Chamberlin, M. E. et al. Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations. Am J Hum Genet 66, 347–355 (2000).
Mudd, S. H. et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 37, 1–31 (1985).
Kozich, V. & Kraus, J. P. Screening for mutations by expressing patient cDNA segments in E. coli: homocystinuria due to cystathionine beta-synthase deficiency. Hum Mutat 1, 113–123 (1992).
de Franchis, R., Kraus, E., Kozich, V., Sebastio, G. & Kraus, J. P. Four novel mutations in the cystathionine beta-synthase gene: effect of a second linked mutation on the severity of the homocystinuric phenotype. Hum Mutat 13, 453–457 (1999).
Gaustadnes, M. et al. The molecular basis of cystathionine beta-synthase deficiency in Australian patients: genotype-phenotype correlations and response to treatment. Hum Mutat 20, 117–126 (2002).
Lee, S. J. et al. Identification and functional analysis of cystathionine beta-synthase gene mutations in patients with homocystinuria. J Hum Genet 50, 648–654 (2005).
Urreizti, R. et al. Spectrum of CBS mutations in 16 homocystinuric patients from the Iberian Peninsula: high prevalence of T191M and absence of I278T or G307S. Hum Mutat 22, 103 (2003).
Katsushima, F. et al. Expression study of mutant cystathionine beta-synthase found in Japanese patients with homocystinuria. Mol Genet Metab 87, 323–328 (2006).
Silao, C. L., Fabella, T. D., Rama, K. I., Estrada, S. C. & Novel, C. B. S. gene mutations in a Filipino patient with Classical Homocystinuria. Pediatr Int, 10.1111/ped.12666 (2015).
El-Said, M. F. et al. A common mutation in the CBS gene explains a high incidence of homocystinuria in the Qatari population. Hum Mutat 27, 719 (2006).
Sebastio, G., Sperandeo, M. P., Panico, M., de Franchis, R., Kraus, J. P. & Andria, G. The molecular basis of homocystinuria due to cystathionine beta-synthase deficiency in Italian families and report of four novel mutations. Am J Hum Genet 56, 1324–1333 (1995).
Yang, Y. et al. Identification of a novel MYOC mutation in a Chinese family with primary open-angle glaucoma. Gene 571, 188–93 (2015).
Martinez-Gutierrez, J. D., Mencia-Gutierrez, E., Gracia-Garcia-Miguel, T., Gutierrez-Diaz, E. & Lopez-Tizon, E. Classical familial homocystinuria in an adult presenting as an isolated lens subluxation. Int Ophthalmol 31, 227–232 (2011).
Cozar, M. et al. Identification and functional analyses of CBS alleles in Spanish and Argentinian homocystinuric patients. Hum Mutat 32, 835–842 (2011).
Munke, M., Kraus, J. P., Ohura, T. & Francke, U. The gene for cystathionine beta-synthase (CBS) maps to the subtelomeric region on human chromosome 21q and to proximal mouse chromosome 17. Am J Hum Genet 42, 550–559 (1988).
Kraus, J. P. et al. The human cystathionine beta-synthase (CBS) gene: complete sequence, alternative splicing and polymorphisms. Genomics 52, 312–324 (1998).
Shan, X. & Kruger, W. D. Correction of disease-causing CBS mutations in yeast. Nat Genet 19, 91–93 (1998).
Kwok, J. S. et al. CBS gene mutations found in a Chinese pyridoxine-responsive homocystinuria patient. Pathology 43, 81–83 (2011).
Kruger, W. D., Wang, L., Jhee, K. H., Singh, R. H. & Elsas, L. J., 2nd . Cystathionine beta-synthase deficiency in Georgia (USA): correlation of clinical and biochemical phenotype with genotype. Hum Mutat 22, 434–441 (2003).
Acknowledgements
This study was supported by grants from the Natural Science Foundation of China (81371048 (B.G.), 81570848 (C.Q.), 81170882 (Y.S.) and 81430008 (Z.Y.)); The Department of Science and Technology of Sichuan Province, China (2015HH0031 (B.G.), 2014JZ0004 (Y.S.), 2011JTD0020 (Z.Y.), 2012SZ0219 (Z.Y.), 2015SZ0052 (Z.Y.), 2014JZ0004 (Y.L.) and 2014JZ0058 (Z.Y.)). We thank all the participants in this study.
Author information
Authors and Affiliations
Contributions
Z.Y. and G.M. designed the study. C.Q., L.L., Z.L., Y.W., G.Z. and Y.X. recruited the participants. B.G., Y.S., X.L., F.H., X.F. and Z.Y. performed the genotyping. B.G. wrote the initial draft. Z.Y., Y.L. and X.L. corrected the English spelling and grammar. All authors critically revised, reviewed and gave final approval of this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gong, B., Liu, L., Li, Z. et al. Novel Compound Heterozygous CBS Mutations Cause Homocystinuria in a Han Chinese Family. Sci Rep 5, 17947 (2015). https://doi.org/10.1038/srep17947
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep17947
This article is cited by
-
Association of selected genetic variants in CBS and MTHFR genes in a cohort of children with homocystinuria in Sri Lanka
Journal of Genetic Engineering and Biotechnology (2022)
-
Mechanisms of Toxic Effects of Homocysteine on the Nervous System
Neurophysiology (2019)
-
Eight novel mutations of CBS gene in nine Chinese patients with classical homocystinuria
World Journal of Pediatrics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
