
Abstract
Hibernation is a physiological adaptation to overcome extreme environmental conditions. It is characterized by prolonged periods of torpor interrupted by temporary arousals during winter. During torpor, body functions are suppressed and restored rapidly to almost pre-hibernation levels during arousal. Although molecular studies have been performed on hibernating rodents and bears, it is unclear how generalizable the results are among hibernating species with different physiology such as bats. As targeted blood proteomic analysis are lacking in small hibernators, we investigated the general plasma proteomic profile of European Myotis myotis and hibernation associated changes between torpid and active individuals by two-dimensional gel electrophoresis. Results revealed an alternation of proteins involved in transport, fuel switching, innate immunity and blood coagulation between the two physiological states. The results suggest that metabolic changes during hibernation are associated with plasma proteomic changes. Further characterization of the proteomic plasma profile identified transport proteins, coagulation proteins and complement factors and detected a high abundance of alpha-fetoprotein. We were able to establish for the first time a basic myotid bat plasma proteomic profile and further demonstrated a modulated protein expression during torpor in Myotis myotis, indicating both novel physiological pathways in bats in general and during hibernation in particular.
Similar content being viewed by others

Introduction
In order to overcome food limitation and high metabolic energy demands during winter periods, animals in temperate climate zones have evolved strategies such as diet switching, annual migration, winter resting or hibernation. Hibernating species undergo a circannual rhythm between homeothermy (activity) and heterothermy (hibernation), in which the heterothermic hibernation cycle alters between extended phases of deep torpor interspersed by short rewarming phases called arousals1. During the torpid phase, which lasts between 6 and 40 days, the metabolic rate drops to 2% of normal coinciding with lowered body temperatures between 10 °C and −2 °C, decreased heart rate and longer breath intervals1,2,3. During arousal, torpor-associated physiological changes are restored to euthermic values for 10–15 hours1. As a consequence of metabolic suppression, the immune system is also functionally suppressed during torpor and restored during arousals in order to clear infections4.
To describe the mechanism underlying the circannual rhythm of hibernation a two-transition model has been proposed, which designates a transition from homeothermy to heterothermy and another cyclic transition within the heterothermic state between torpor and arousal5,6. Although the precise genetic and molecular regulation underlying this mechanism remains unclear, it was shown that the switch from homeothermy to heterothermy is associated with a broad differential expression of existing genes rather than with the evolution and expression of hibernation-specific genes7,8,9. These expression differences occur at the protein level in heart10, intestine11, liver12 and skeletal muscles13 of ground squirrels (Ictidomys tridecemlineatus). In these tissues, increased expression levels of proteins involved in glycolysis, glycogenesis and amino acid catabolism were observed during the active state, while hibernation was characterized by increased expression of proteins involved in fatty acid catabolism. These findings were consistent with the shift from carbohydrates to lipid oxidation during hibernation14. Another study investigating the kidney proteome in the same species found evidence for a turnover of plasma proteins alpha-2-macroglobulin, albumin and apolipoprotein during torpor-arousal cycles15.
Plasma proteins and their composition in the blood are known to be an important indicator of physiological changes including disease recognition or pathogen infection in humans16. Thus hibernation-associated changes in blood plasma protein composition may also be detectable. In Asian chipmunks (Tamias sibiricus asiaticus), for example, specific proteins associated with hibernation have been identified in plasma17,18, while in ground squirrels (Ictidomys tridecemlineatus), altered plasma metabolites (e.g. amino acids, regulatory lipids) during the heterothermic hibernation cycle were characterized using non-targeted metabolomic approaches19,20. A targeted proteomic analysis of the blood serum in hibernating American black bears (Ursus americanus) revealed differential expression of proteins involved in immunity, coagulation and bone metabolism21. These results provide molecular support for the peculiarities of ursid hibernation, including wound healing and active immune function during hibernation22. In contrast to bears, rodent hibernation is associated with immunosuppression4, highlighting the importance of research on blood proteomics in small mammalian hibernators given the lack of a universal mammalian hibernation proteomic profile.
Research on mammalian hibernation has focused on rodents. Although there is information available on general regulatory processes from tissue proteomic studies in rodents, targeted analysis of plasma and serum proteome profiles is lacking. Even if the hibernating phenotype between species is similar across taxa, differences in the mechanisms leading to hibernation associated changes are unclear, especially if physiological adaptations during homeothermy are considered. Accordingly, similarities in physiological aspects of hibernation may be expected for rodents and bats, e.g. fuel change from carbohydrate to lipid metabolism, reduction in protein synthesis, among others. However other processes may be unique due to basic differences in their physiology. Bats are able to enter a heterothermic state throughout the year by entering bouts of prolonged torpor also including the summer period23 while most hibernating rodents exhibit heterothermy only during the winter season. Bats are also the only mammalian group capable of powered flight, which has selected for increased metabolic capacity and elevated antioxidant levels24. Moreover, they are unusually long-lived species25, traits which are different from the hibernating rodents studied to date. Furthermore, the reproduction patterns of hibernating bats26,27 or the role of wing membrane in maintaining the water balance28, makes chiropteran hibernation related physiology unique. It has been suggested that some bat species are not immunosuppressed during hibernation as observed in rodents and that bats maintain specific defenses against psychrophilic pathogens such as Pseudogymnoascus destructans29. Therefore, we hypothesized that some of the regulatory mechanisms of hibernating bats should be distinct from those of rodents.
In order to improve our knowledge on chiropteran hibernation and in small mammalian hibernation in general, we compared the blood plasma proteomic profile of a hibernating European bat species, the greater mouse-eared bat (Myotis myotis) by using a two-dimensional gel electrophoresis approach to characterize differential expressed proteins between homeothermic and heterothermic, torpid individuals. In comparison to rodent hibernation we predict for bats similar pattern of protein expression in general regulatory mechanisms but differences in the regulation of proteins involved in specific physiological processes such as immune function or reproduction.
Materials and Methods
Ethics statement
All experimental procedures described in the materials and methods section were approved by the animal welfare and ethics committee of the Leibniz Institute for Zoo and Wildlife Research (permit #2010-05-01). All experiments were carried out in accordance with the approved guidelines of the Leibniz Institute for Zoo and Wildlife Research.
Sample collection
Greater mouse-eared bats (M. myotis) were captured in Northern Bavaria (Germany), under the license of regional governments (permits 54-2532.2-9/10, 55.1-8642G062/10 and 55.1-8642-01-17/10). Blood samples from 14 male individuals in the homeothermic active state (n = 7) were captured in September 2010 and torpid individuals (n = 7) during the hibernation period in March 2011. Active individuals were captured using mistnets (Ecotone, Poland), while torpid individuals were picked by hand from the walls of the hibernacula. Blood samples were collected in active bats from the uropatagial vein using sterile needles and by transferring blood droplets into heparinized microcapillary tubes and in torpid bats from the jugular vein using a sterile heparinized needle and syringe. All bats were released at the site of capture after bleeding was completed. Structural and functional immunological measurements were performed on fresh blood while in case of surplus blood the plasma was separated by centrifugation and stored at −80 °C until further analysis.
2-Dimensional Fluorescence Difference Gel Electrophoresis (2-D DIGE)
Proteomic profiles were determined from blood plasma samples of 14 M. myotis male individuals (7 homeothermic, active individuals and 7 heterothermic, torpid individuals) using 2-D DIGE.
Serum albumin is the most abundant plasma protein in humans16 and can impede the detection and quantification of low abundance plasma proteins30. Therefore plasma investigation studies often deplete albumin prior to analysis. However the depletion of albumin also can remove untargeted proteins31. Albumin depletion was not performed in this study but serum albumin was excluded during mass spectrometry identification.
Total plasma protein concentration was determined using a NanoDrop® and diluted to the required concentration of 0.55 μg protein/μL in labeling buffer [50 mM tris, 5 mM EDTA, 5% v/v glycerol, pH 7.2; final volume = 9 μL] for fluorescent protein labeling using S-Dye300 of the Saturn-2DTM labeling kit (NH DyeAGNOSTICS GmbH, Germany) according to manufacturer´s protocol. An internal standard (IS) consisting of all samples used in each experimental procedure was diluted to the required concentration of 0.55 μg protein/μL in labeling buffer (final volume = 9 μL) and fluorescent labeled using S-Dye200 of the Saturn-2DTM labeling kit.
Labeled samples (per gel: 9µL of an individual S-Dye300-labeled sample + 9µL of S-Dye200-labeled IS) were diluted in 432 µL rehydration buffer [8 M Urea, 1% w/v 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS), 13 mM Dithiothreitol (DTT), 0.5% v/v Servalyt (SERVA Electrophoresis GmbH, Germany)] and loaded on IPG BlueStrips pH 3–10 NL/24 cm (SERVA Electrophoresis GmbH, Germany) for active (50 V, 15 h) sample-in-gel rehydration using PROTEAN® IEF Cell tray (Bio-Rad, USA). Isoelectric focusing was performed under following conditions: step 1, 300 V, 150 V/h rapid; step 2, 600 V, 300 V/h rapid; step 3, 1500 V, 750 V/h rapid; step 4, 3000 V, 48000 V/h rapid; step 5, 6000 V, 10000 V/h rapid; step 6, 300 V, 5 h; total 60700 V/h.
Prior to second dimension separation, IPG stripes were equilibrated in equilibration buffer [EB: 6 M Urea, 2% SDS, 0.375 M Tris, 20% v/v glycerol] with first 20 mg/mL DTT for 15 min, followed by EB with 25 mg/mL iodoacetamide (IAA) for 15 min. After equilibration, stripes were placed on 15% SDS gels in 27.5 × 22 cm low fluorescence glass cassettes (NH DyeAGNOSTICS GmbH, Germany) and overlaid with 1% agarose including bromphenol blue. Gel electrophoresis was performed in a SE900 electrophoresis unit (Hoefer Inc., USA) for a minimum of 1900 V/hours and a maximum of 2200 V/hours at 80 mA/ gel, 100 W and 100 V. Imaging of the gels was performed by fluorescence scanning on a Typhoon 9400 Imager (GE Healthcare, USA) at excitation/emission wavelengths of 532/576 nm (S-Dye200) and 633/664 nm (S-Dye300).
To evaluate the expression pattern of protein spots separated by 2-D DIGE, all sample gels were analyzed using the Delta2D software (DECODON, Germany). An IS S-Dye200 image was designated as the master gel based on the largest number of detectable spots and then connected to all images by a “sample in gel” warping strategy in the Delta2D software. Warping of gels was done by defining matched vectors between distinct protein spots chosen automatically and manually. For expression analysis of protein spots, a fused image of all sample images (S-Dye300; gel images of each sample are shown in supplementary Figure S1) was generated and a consensus spot pattern for normalization against IS images was applied. Matched protein spots present in all sample images and with a minimum of 1.5 fold change between active and torpid state were statistically analysed using a non-parametric Wilcoxon Rank Sum test (alpha: p < 0.05) with the Delta2D statistic software TMeV (Decodon).
Preparative 2-D gel for protein identification
Preparative gel separation was employed using pooled samples of all 14 individuals. For the first dimension, unlabeled pooled plasma (total protein concentration = 240 μg) was loaded onto an IPG stripe and separated according to isoelectric points as described above. Separation according to molecular weight in the second dimension was also performed as described above with the exception that a 28 × 21 cm hinged glass cassette (Hoefer Inc., USA) was used instead of low fluorescence glass cassettes. After 2-D gel electrophoresis the gel was Coomassie blue dye stained [0.02% Coomassie blue G-250; 5% w/v aluminum sulfate; 10% v/v ethanol; 2% v/v ortho-phosphoric acid in dH2O] for 4 h and then destained [10% ethanol; 2% v/v ortho-phosphoric acid in dH2O] to remove unbound Coomassie blue32. Spots with a fold change difference of ≥1.5 and a statistical significance of p < 0.05 between the active and torpid state were picked for protein identification. It was not possible to pick all differentially expressed spots as not all spots were distinguishable on the Coomassie blue stained gel. Additional spots for a basic survey of the bat plasma proteome were chosen based on a distinct appearance in the preparative gel and co-localization with protein spots that were differentially expressed. Protein spots of interest were picked manually and stored in Eppendorf tubes containing 5% v/v acetic acid (in dH2O) at 4 °C until analysed by mass spectrometry.
Protein identification by mass spectrometry (MS)
Excised gel spots were washed with water, 25 mM ammonium bicarbonate in acetonitrile/water (1:1) and 50 mM ammonium bicarbonate, shrunk by dehydration in acetonitrile and dried in a speed-vacuum centrifuge. The dry gel pieces were re-hydrated in 20 μL of 50 mM ammonium bicarbonate containing 50 ng trypsin (sequencing grade modified, Promega). After incubation at 37 °C overnight, the enzymatic reaction was terminated by addition of 20 μL of 0.5% (v/v) trifluoroacetic acid in acetonitrile, the liquid was separated, evaporated to dryness under vacuum and the tryptic peptides were re-dissolved in 6 μL 0.1% (v/v) trifluoroacetic acid , 5% (v/v) acetonitrile in water.
MALDI mass spectrometry was performed as previously described33. In brief, the peptides were purified on a C18 RP minicolumn (ZipTip C18, Millipore, Bedford, MA) and eluted directly onto the MALDI target plate using alpha-cyano-hydroxycinnamic acid matrix solution. MS and MS/MS measurements were performed using a MALDI-TOF-TOF instrument (AB SCIEX TOF/TOF 5800; Applied Biosystems, Framingham, MA, USA) equipped with a Neodymium-doped yttrium lithium fluoride laser (Nd:YLF, 349 nm). MS spectra were acquired in positive ion reflector mode by accumulating 5000 consecutive laser shots. For MS/MS, a maximum of 20 precursor ions were selected automatically. GPS Explorer (version 3.6, Applied Biosystems) was used to process the spectra.
LC-MS/MS analyses were performed on an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher) equipped with an Ultimate 3000 nanoLC system (Thermo Scientific). For separation of tryptic peptides, a capillary column (PepMap100, C18, 3 μm, 100 Å, 250 mm × 75 μm i.d., Thermo Scientific) was used. Elution was performed at a flow rate of 300 nL/min using a gradient of 3–50% B in 30 min. Mobile phase A contained 0.1% formic acid in water and mobile phase B contained 0.1% formic acid in acetonitrile. Mass spectra were acquired in a data-dependent mode with one MS survey scan (with a resolution of 60,000) in the Orbitrap and MS/MS scans of the five most intense precursor ions in the linear trap quadrupole. The dynamic exclusion time for precursor ions was set to 90 s and automatic gain control was set to 1 × 106 for Orbitrap-MS and 10,000 for LTQ-MS/MS scans. The Mascot Distiller Quantitation Toolbox (Matrix Science) was used to generate peak lists.
The processed MS data were analysed on a MASCOT (mass spectral search algorithm) server (version 2.2.2, Matrix Science Ltd, London) and searched in-house against the mammalian subset of the NCBI database (version 221013; 33,055,681 sequences). For MALDI-MS, the mass tolerance of precursor and sequence ions was set to 100 ppm and 0.35 Da, respectively. For LC-MS/MS, the mass tolerance of precursor and sequence ions was set to 10 ppm and 0.35 Da, respectively. A maximum of two missed cleavages was allowed. Methionine oxidation and the acrylamide modification of cysteine were used as variable modifications. A protein was accepted as identified if the total MASCOT score was greater than the significance threshold and at least two peptides appeared the first time in the report and were the top ranking peptides (peptide matches of all proteins identified are listed in supplementary Table S2). For MALDI data, the protein score was −10*log(p), where p is the probability that the observed match is a random event, e.g. protein scores greater than 75 are significant(p < 0.05). For LC-MS/MS data, the ions score was −10*log(p), where p is the probability that the observed match is a random event, e.g. individual ions scores >41 indicate identity or extensive homology (p < 0.05).
Results
Differential protein expression of plasma proteins in M. myotis
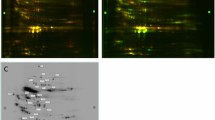
When considering all gel images, we detected a total of 204 matched protein spots present in all individual samples by using a consensus spot pattern. Of 204 protein spots, 13 protein spots (6.4%) showed a significant (p < 0.05) differential expression with a minimum of a 1.5 fold difference between the two physiological states, nine protein spots being down regulated and four being up regulated in torpor compared to the active state (Fig. 1 and Table 1).

Myotis myotis plasma proteome.
Fused image of representative images of S-dye labeled M. myotis active and torpid plasma samples separated by 2-DIGE. Differential expressed protein spots (p < 0.05; minimum fold-change 1.5) are displayed in either green for up regulated or red for down regulated proteins during torpor and indicated by numbers corresponding to Table 1. Protein spots which were identified via mass spectrometry are underlined. The IPG stripe pH range is indicated on top of the image.
We were not able to determine and pick all differential expressed protein spots on the Coomassie stained gel. Protein identification (IDs) were successful for 5 out of 13 differential expressed protein spots (S1/C7, S4/C15, S5/C16, S6/C17 down regulated; S2/C3 up regulated), in which MS data obtained multiple protein IDs for 3 protein spots (S1/C7, S5/C16, S6/C17) and single protein IDs for 2 protein spots (S2/C3, S4/C15). For multiple protein IDs, the top 3 ranked MS peptide matches are shown in Table 1, excluding matches to serum albumin.
Protein matches to those identified in Myotis davidii and Myotis brandtii based on a MASCOT search of the NCBI database. The significantly down regulated proteins during the torpid state were identified as vitamin D-binding protein, vascular non-inflammatory molecule 3, anti-thrombin III (multiple protein IDs of spot S1/C7), serotransferrin (S4/C15; S5/C16; S6/C17), ß-chain of fibrinogen (S6/C17) and alpha-fetoprotein (S5/C16). The significantly up regulated protein in torpor (S2/C3) was identified as Kininogen-1 (Table 1).
Survey of expressed plasma proteins in M. myotis
To describe a general proteomic profile of bat plasma, 27 protein spots co-localizing with differentially expressed protein spots were picked for analysis (Fig. 2). For 18 out of 27 picked protein spots, MS data yielded multiple protein IDs. The top 3 ranks of MS peptide match results are shown in Table 2 excluding matches to serum albumin. Identified proteins matched those identified in Myotis davidii and Myotis brandtii based on MASCOT searches of the NCBI protein database. In total, MS yielded 20 different protein IDs of which 12 protein IDs appeared in more than one protein spot. Protein IDs appearing in numerous protein spots included alpha-fetoprotein (20 protein spots (PS)), serotransferrin (7 PS), anti-thrombin III (6 PS), vitamin D-binding protein (6 PS), hemoglobin subunit beta (5 PS), complement C4A (3 PS), fibrinogen beta chain (3 PS), vascular non-inflammatory molecule 3 (3 PS), alpha-1-antitrypsin (2 PS), hemoglobin subunit alpha (2 PS), hemoglobin alpha chain (2 PS) and kininogen-1 (2 PS). Protein IDs found in single protein spots were kininogen-2 (C1), complement C3 (C13), NSFL1 cofactor p47 (C18), apolipoprotein A-V (C19), chain A profilin-Beta-Actin (C20), carbonic anhydrase 2 (C24), adenine phosphoribosyltransferase (C26), apolipoprotein M (C29) and dihydroorotate dehydrogenase (C30) (Table 2). Protein IDs appearing in more than one protein spot were located in similar gel regions (C2, C4, C6, C8 and C9; C11 and C12) expect protein IDs alpha-fetoprotein, fibrinogen, serotransferrin and hemoglobin subunit beta which were located in protein spots distributed over all gel regions.

Coomassie Gel of Myotis myotis plasma proteome.
Preparative Coomassie gel of all 14 plasma samples (pooled) separated by two-dimensional gel electrophoresis. The IPG stripe pH range is indicated on top of the image. Protein numbers display spots that were picked based on co-localization with hibernation specific differentially expressed protein spots distinct appearance in the preparative gel.
Discussion
We observed differential expression for 13 of 204 detectable spots between the active and torpid state representing 6.4% of the total detected protein spots, a similar proportion observed by targeted serum proteomic analysis in American black bears21. The protein gel spot pattern exhibited electrophoretic accumulation of protein spots, suggesting an occurrence of multiple protein isoforms. The plasma protein signature clearly differentiated the torpid state from the active homeothermy which is consistent with observed variation in tissues of other hibernating mammalian species10,11,12,13,14. Mass spectrometric analysis identified seven differentially regulated proteins. Most of them were down regulated (Vitamin D-binding protein (DBP), Serotransferrin (TF), Vascular non inflammatory molecule 3 (VANIN-3), Alpha-fetoprotein (AFP), Fibrinogen beta chain (FIG-β) and Anti-thrombin III (AT)), whereas Kininogen-1 (KNG1) was up regulated.
Down regulated proteins TF, AFP and DBP are classified as transport proteins based on their primary function of binding essential body metabolites, vitamins or metal ions. The iron binding glycoprotein TF plays an important role in cellular and systemic iron homeostasis particularly during long fasting periods34. DBP and AFP belong to the albumin gene family in humans and have multifunctional roles in plasma. DBP is a major transporter of Vitamin D3 and its metabolites and is found as a free plasma protein and also on the surface of many cell types including blood cells35,36. In contrast, AFP occurs primarily as a free plasma protein in numerous polymeric forms37. AFP is involved in the binding and transport of several metabolites including fatty acids, bilirubin and estrogens or metal ions in human fetuses and other mammals37,38,39. The down regulation of these transport proteins might reflect the decreased metabolic rate of hibernating species3. Consistent with this hypothesis, studies on diet-restricted rats demonstrated lower mRNA levels of DBP and a decrease of expressed DBP in the liver during fasting40,41. Furthermore TF and DBP are involved in the modulation of innate immunity42,43,44 and DBP has a particularly important role in the activation of macrophages45. Therefore, down regulation suggests a reduction of innate immunity during hibernation4.
VANIN-3 is an amidohydrolase involved in the catabolism of CoenzymeA (CoA) from pantothenic acid (Vitamin B5). CoA and its thioester form acetyl-CoA, are essential cofactors in maintaining fatty acid balance46. During hibernation, a switch from glycolysis to the oxidation of triacylglycerols is observed14 making fatty acids of adipose tissue the primary energy source. However, the function of VANIN-3 in CoA catabolism is poorly understood. Down regulation during hibernation might suggest an alternate catabolism of CoA reflecting the switch from glucose to triacylglycerols as an energy source.
Differentially expressed proteins FIG-β, AT and KNG1 are part of the blood coagulation system. FIG-β together with other protein domains forms the soluble glycoprotein fibrinogen which is converted by thrombin into insoluble fibrin during formation of blood clots47. The serine protease inhibitor AT degrades proteases of the coagulation cascade in order to regulate coagulation and to prevent thrombosis48. KNG1 in contrast is part of the kallikrein-kinin system and is known to be essential in many pathways including thrombosis, vascular permeability and inflammation49. During hibernation, platelet aggregation is reduced in brown bears (Ursus arctos)50. Also an elevation of the protease inhibitor alpha-2-macroglobulin in serum of hibernating ground squirrels and black bears was observed, consistent with reduced coagulation21,51. In addition, KNG1 was found to be down regulated in hibernating black bears21. A decrease of coagulation activity during hibernation could protect the individual from blood clotting during periods of low heart rate and reduced blood flow1,52. However, the expression pattern of down regulated AT and up regulated KNG1 in M. myotis could suggest an elevated coagulation capacity. Similarly, up regulation of coagulation associated genes have been described in hibernating Myotis brandtii at the transcription level25. In contrast a down regulation of FIG-β during torpor in M. myotis may result in a reduction of coagulation capacity53. Based on the contradictory findings of proteins involved in coagulation in M. myotis we conclude that the detected coagulation associated proteins might be involved in other physiological processes relevant to hibernation. This may imply that bats have a different coagulation cascade compared to other mammals as it is unlikely that increased coagulation during hibernation would be advantageous.
In order to establish a basic plasma proteomic profile in bats, twenty-seven clearly definable protein spots co-localizing with hibernation specific differentially expressed protein spots were identified. MS protein identification yielded multiple IDs per protein spot (Table 2) and IDs found in more than one protein spot including differential expressed protein spots suggesting the co-occurrence of different isoforms, which can be regulated differentially54. Most proteins identified are known to be the most abundant proteins in human plasma16 including TF, FIG-β, alpha-1-antitrypsin and complement C3. Based on their primary functions, further identified proteins could be categorized as transport proteins (e.g. hemoglobin, apolipoprotein A-V and M), coagulation proteins (e.g. AT, kininogen I and II), proteins of the complement system (e.g. complement factor C4A and C3) and proteins involved in a variety of regulative processes (e.g. carbonic anhydrase). All identified proteins in M. myotis can be found in a general plasma profile of humans16 indicating similarities in the plasma protein composition across mammalian taxa. However, AFP exhibited an unusual profile by identification in 21 of 34 picked protein spots suggesting a high abundance in plasma of adult M. myotis. In humans, AFP is the most abundant protein during fetal development and has a similar function to serum albumin in adults including the binding of hydrophobic ligands such as fatty acids, metabolites and as well metal ions35,36,37. The abundance of AFP in the adult bat plasma suggests that this protein may play an important functional role in different mechanisms in M. myotis, maybe in bats in general, thus deserving further scientific attention.
Protein identification was hindered by the lack of protein databases for non-model, wildlife species. Homologous proteins of other species do not always share 100% sequence similarity with unknown M. myotis proteins, reducing the amount of potential analysable tryptic peptides per protein depending on the level of similarity which influences the peptide match ranking. This lack of knowledge will certainly change as proteomic research on non-model species develops further55. Nonetheless, we were able to establish a basic plasma proteome profile for M. myotis. Moreover, we showed differential expression of plasma proteins in hibernating bats compared to active bats demonstrating a modulation of proteins involved in transport, fuel switching from carbohydrate to triacylglycerol oxidation, innate immunity and blood coagulation cascade. In addition protein identification of further protein spots demonstrated evidence for an alternate composition of high abundant plasma proteins in M. myotis with AFP as a possible prominent protein in adult bat plasma. Chiropteran hibernation proteome profile was generally consistent with other hibernating species at the pathway level as hypothesized except for coagulation which appears to be myotid bat specific. Further comparisons with rodents and bears will clarify the general similarities and differences among hibernating species proteomic profiles particularly as protein databases improve and identification becomes facilitated.
Additional Information
How to cite this article: Hecht, A. M. et al. Plasma proteomic analysis of active and torpid greater mouse-eared bats (Myotis myotis). Sci. Rep. 5, 16604; doi: 10.1038/srep16604 (2015).
References
Carey, H. V., Andrews, M. T. & Martin, S. L. Mammalian hibernation: cellular and molecular responses to depressed metabolism and low temperature. Physiol. Rev. 83, 1153–1181 (2003).
Barnes, B. M. Freeze avoidance in a mammal: body temperatures below 0°C in an Arctic hibernator. Science. 244, 1593–1595 (1989).
Geiser, F. Metabolic rate and body temperature reduction during hibernation and daily torpor. Annu. Rev. Physiol. 66, 239–274 (2004).
Bouma, H. R., Carey, H. V. & Kroese, F. G. Hibernation: the immune system at rest? J. Leukoc. Biol. 88, 619–624 (2010).
Serkova, N. J., Rose, J. C., Epperson, L. M., Carey, H. V. & Martin, S. L. Quantitative analysis of liver metabolites in three stages of the circannual hibernation cycle in 13-lined ground squirrels by NMR. Physiol. Genomics. 31, 15–24 (2007).
Hampton, M. & Andrews, M. T. A simple molecular mathematical model of mammalian hibernation. J. Theor. Biol. 247, 297–302 (2007).
Srere, H. K., Wang, L. C. & Martin, S. L. Central role for differential gene expression in mammalian hibernation. Proc. Natl. Acad. Sci. USA. 89, 7119–7123 (1992).
Boyer, B. B. & Barnes, B. M. Molecular and metabolic aspects of mammalian hibernation. Bioscience. 49, 713–724 (1999).
Yan, J., Barnes, B. M., Kohl, F. & Marr, T. G. Modulation of gene expression in hibernating arctic ground squirrels. Physiol. Genomics. 32, 170–181 (2007).
Grabek, K. R. et al. Multistate proteomics analysis reveals novel strategies used by a hibernator to precondition the heart and conserve ATP for winter heterothermy. Physiol. Genomics. 43, 1263–1275 (2011).
Martin, S. L. et al. Proteomic analysis of the winter-protected phenotype of hibernating ground squirrel intestine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R316–R328 (2008).
Epperson, L. E., Rose, J. C., Carey, H. V. & Martin, S. L. Seasonal proteomic changes reveal molecular adaptations to preserve and replenish liver proteins during ground squirrel hibernation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R329–R340 (2010).
Hindle, A. G., Karimpour-Fard, A., Epperson, L. E., Hunter, L. E. & Martin, S. L. Skeletal muscle proteomics: carbohydrate metabolism oscillates with seasonal and torpor-arousal physiology of hibernation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1440–R1452 (2011).
Schwartz, C. & Andrews, M. T. Cirannual transitions in gene expression: Lessons from seasonal adaptations. Curr. Top. Dev. Biol. 105, 247-273 (2013).
Jani, A. et al. Kidney proteome changes provide evidence for a dynamic metabolism and regional redistribution of plasma proteins during torpor-arousal cycles of hibernation. Physiol. Genomics. 44, 717–727 (2012).
Anderson, N. L. et al. The human plasma proteome: A nonredundant list developed by combination of four separate sources. Mol. Cell. Proteomics. 3, 311–326 (2004).
Kondo, N. & Kondo, J. Identification of novel blood proteins specific for mammalian hibernation. J. Biol. Chem. 267, 473–478 (1992).
Takamatsu, N., Ohba, K. I., Kondo, J., Kondo, N. & Shiba, T. Hibernation-associated gene regulation of plasma proteins with a collagen-like domain in mammalian hibernators. Mol. Cell. Biol. 13, 1516–1521 (1993).
Nelson, C. J., Otis, J. P. & Carey, H. V. Global analysis of circulating metabolites in hibernating ground squirrels. Comp. Biochem. Physiol. D. 5, 265–273 (2010).
Epperson, L. E., Karimpour-Fard, A., Hunter, L. E. & Martin, S. L. Metabolic cycles in a circannual hibernator. Physiol. Genomics. 43, 799–807 (2011).
Chow, B. A., Donahue, S. W., Vaughan, M. R., McConkey, B. & Vijayan, M. M. Serum immune-related proteins are differentially expressed during hibernation in the American black bear. PLoS ONE. 8, e66119 (2013).
Iaizzo, P. A., Laske, T. G., Harlow, H. J., McCLAY, C. B. & Garshelis, D. L. Wound healing during hibernation by black bears (Ursus americanus) in the wild: elicitation of reduced scar formation. Integr. Zool. 7, 48–60 (2012).
Wojciechowski, M. S., Jefimow, M. & Tegowska E. Environmental conditions, rather than season, determine torpor use and temperature selection in large mouse-eared bats (Myotis myotis). Comp. Biochem. Physiol., Part A Mol. Integr. Physiol. 147, 828–840 (2007).
Zhang, G. et al. Comparative analysis of bat genomes provides insight into the evolution of flight and immunity. Science. 339, 456–460 (2013).
Seim, I. et al. Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nat. Commun. 4, 2212 doi: 10.1038/ncomms3212 (2013).
Oxberry, B. A. Female reproductive patterns in hibernating bats. J. Reprod. Fert. 56, 359–367 (1979).
Gustafson, A. W. Male reproductive patterns in hibernating bats. J. Reprod. Fert. 56, 317–331 (1979).
Cryan, P. M., Meteyer, C. U., Boyles, J. G. & Blehert, D. S. Wing pathology of white-nose syndrome in bats suggests life-threatening disruption of physiology. BMC Biology. 8, 135 (2010).
Puechmaille, S. J. et al. White-nose syndrome: is this emerging disease a threat to European bats? Trends Ecol. Evol. 26, 570–576 (2011).
Tu, C. et al. Depletion of abundant plasma proteins and limitations of plasma proteomics. J. Proteome Res. 9, 4982–4991 (2010).
Zolotarjova, N. et al. Differences among techniques for high-abundant protein depletion. Proteomics. 5, 3304–3313 (2005).
Dyballa, N. & Metzger, S. Fast and sensitive colloidal coomassie G-250 staining for proteins in polyacrylamide gels. JoVE. 30, 1431 (2009).
Strathman, J. et al. Proteome analysis of chemically induced mouse liver tumors with different genotype. Proteomics. 7, 3318–3331 (2007).
Scriba, P. C. et al. Effects of obesity, total fasting and re-alimentation on I-tyroxine (T4), 3, 5, 3′-l-triiodothronine (T3), 3, 3′, 5′-l-triiodothronine (rT3), tyroxine binding globulin (TBG), cortisol, thyrothrophin, cortisol binding globulin (CBG), transferrin, α2-haptoglobin and complement C3 in serum. Acta Endocrinol. 91, 629–643 (1979).
White, P. & Cooke, N. The multifunctional properties and characteristics of vitamin D-binding protein. Trends Endocrin. Met. 11, 320–327 (2000).
Gomme, P. T. & Bertolini, J. Therapeutic potential of vitamin D-binding protein. Trends Biotechnol. 22, 340–345 (2004).
Terentiev, A. A. & Moldogazieva, N. T. Alpha-fetoprotein: a renaissance. Tumor Biol. 34, 2075–2091 (2013).
Ruoslahti, E., Ester, T. & Seppala, M. Binding of bilirubin by bovine and human alpha-fetoprotein. Biochim. Biophys. Acta. 578, 511–519 (1979).
Aoyagi, Y., Ikenaka, T. & Ishida, F. Copper (II)-binding ability of human alpha-fetoprotein. Cancer Res. 38, 3483–3486 (1978).
Bertile, F., Schaeffer, C., Le Maho, Y., Raclot, T. & Van Dorsselaer, A. A proteomic approach to identify differentially expressed plasma proteins between the fed and prolonged fasted states. Proteomics. 9, 148–158 (2009).
Laing, C. J. & Fraser, D. R. Changes with malnutrition in the concentration of plasma vitamin D binding protein in growing rats. Br. J. Nutr. 88, 133–139 (2002).
Ponka, P., Beaumont, C. & Richardson, D. R. Function and regulation of transferrin and ferritin. Sem. Hematol. 35, 35–54 (1998).
White, P. et al. The absence of vitamin D binding protein (DBP) slows immune responses during Leishmania donovani infection in vivo. FASEB. 14, A1031: 91.23 (2000).
Kew, R. R. & Webster, R. O. Gc globulin (vitamin D-binding protein) enhances the neutrophil chemotactic activity of C5a and C5a. J. Clin. Invest. 82, 364–369 (1988).
Gavison, R. I. V. K. A. & Bar-Shavit, Z. Impaired macrophage activation in vitamin D3 deficiency: differential in vitro effects of 1, 25-dihydroxyvitamin D3 on mouse peritoneal macrophage functions. J. Immunol. 143, 3686–3690 (1989).
Leonardi, R., Zhang, Y. M., Rock, C. O. & Jackowski, S. Coenzyme A: back in action. Prog. Lipid Res. 44, 125–153 (2005).
Mosesson, M. W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 3, 1894–1904 (2005).
Jesty, J., Lorenz, A., Rodriguez, J. & Wun, T. C. Initiation of tissue factor pathway of coagulation in the presence of heparin: control by antithrombin III and tissue factor pathway inhibitor. Blood. 87, 2301–2307 (1996).
Schmaier, A. H. & McCrae, K. R. The plasma kallikrein-kinin system: its evolution from contact activation. J. Thromb. Haemost. 5, 2323–2329 (2007).
Fröbert, O. et al. Platelet function in brown bear (Ursus arctos) compared to man. Thromb. J. 8, 11 doi: 10.1186/1477-9560-8-11 (2010).
Srere, H. K., Belke, D., Wang, L. & Martin, S. L. a2-Macroglobulin gene expression during hibernation in ground squirrels is independent of acute phase response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 268, R1507–R1512 (1995).
Hellgren, E. C. Physiology of hibernation in bears. Ursus. 10, 467–477 (1998).
Hamaguchi, M., Bunce, L. A., Sporn, L. A. & Francis, C. W. Spreading of platelets on fibrin is mediated by the amino terminus of the beta chain including peptide beta 15-42. Blood. 81, 2348–2356 (1993).
Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470–476 (2008).
Valcu, C. M. & Kempenaers, B. Proteomics in behavioral ecology. Behav. Ecol. 26, 1–15 (2015).
Acknowledgements
We are grateful to Simon Ghanem, Matthias Hammer, Daniel Lewanzik, Katja Pohle, Zaida Renteria-Solis, Jaap van Schaik, Karin Schneeberger, Bernhard Walk and Gudrun Wibbelt for their help during fieldwork and sample collection. Furthermore we would like to thank Angelika Bondzio and Ralf Einspanier for their help with the imaging and Jörns Fickel for critical reading of early drafts of the manuscript. The comments received from two anonymous referees greatly improved the quality of the present work. The research was supported by funds from the International Max Planck Research School for Infectious Diseases and Immunology and from the Leibniz Institute for Zoo and Wildlife Research, Berlin.
Author information
Authors and Affiliations
Contributions
G.Á.C. conceived the study; A.M.H. performed all laboratory experiments expect MS under the supervision of B.C.B. and E.K. performed the MS experiments; A.M.H., E.K. and B.C.B analysed the data; C.C.V. worked on the permissions and collected together with G.Á.C the samples; A.M.H., B.C.B., E.K., A.D.G. and G.Á.C. discussed the results and wrote the manuscript. All authors commented, read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hecht, A., Braun, B., Krause, E. et al. Plasma proteomic analysis of active and torpid greater mouse-eared bats (Myotis myotis). Sci Rep 5, 16604 (2015). https://doi.org/10.1038/srep16604
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16604
This article is cited by
-
Differences in acute phase response to bacterial, fungal and viral antigens in greater mouse-eared bats (Myotis myotis)
Scientific Reports (2022)
-
Phagocyte activity reflects mammalian homeo- and hetero-thermic physiological states
BMC Veterinary Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}