
Abstract
Whilst common genetic variation in many non-coding genomic regulatory regions are known to impart risk of colorectal cancer (CRC), much of the heritability of CRC remains unexplained. To examine the role of recurrent coding sequence variation in CRC aetiology, we genotyped 12,638 CRCs cases and 29,045 controls from six European populations. Single-variant analysis identified a coding variant (rs3184504) in SH2B3 (12q24) associated with CRC risk (OR = 1.08, P = 3.9 × 10−7) and novel damaging coding variants in 3 genes previously tagged by GWAS efforts; rs16888728 (8q24) in UTP23 (OR = 1.15, P = 1.4 × 10−7); rs6580742 and rs12303082 (12q13) in FAM186A (OR = 1.11, P = 1.2 × 10−7 and OR = 1.09, P = 7.4 × 10−8); rs1129406 (12q13) in ATF1 (OR = 1.11, P = 8.3 × 10−9), all reaching exome-wide significance levels. Gene based tests identified associations between CRC and PCDHGA genes (P < 2.90 × 10−6). We found an excess of rare, damaging variants in base-excision (P = 2.4 × 10−4) and DNA mismatch repair genes (P = 6.1 × 10−4) consistent with a recessive mode of inheritance. This study comprehensively explores the contribution of coding sequence variation to CRC risk, identifying associations with coding variation in 4 genes and PCDHG gene cluster and several candidate recessive alleles. However, these findings suggest that recurrent, low-frequency coding variants account for a minority of the unexplained heritability of CRC.
Similar content being viewed by others

Introduction
Heritable factors are thought to contribute to around 35% of the variation in risk of developing colorectal Cancer (CRC)1,2,3. High-penetrance mutations responsible for Mendelian disorders such as Lynch Syndrome, familial adenomatous polyposis and MUTYH associated polyposis have been shown to account for around 5% of all CRC. Genome-wide association studies (GWAS) have vindicated the notion that common genetic variants also contribute to CRC risk. Over 25 risk SNPs identified through GWAS4,5,6,7,8,9,10,11,12,13,14,15 are collectively responsible for only around 1% of CRC heritability3 and so much of the genetic contribution to CRC risk currently remains enigmatic. It has been proposed that low frequency variants in coding regions, may have substantial effects on risk and so may explain an appreciable proportion of the heritability of complex disease16. Conventional GWAS arrays have been sub-optimally configured to genotype such low frequency recurrent variation, whilst large-scale sequencing has been constrained by cost and data analysis bottlenecks.
Exome sequencing studies in multiple populations have enabled the assembly of catalogues of well-characterised single nucleotide variants within the coding sequence of genes. Genotyping arrays have been formatted into “exon” arrays specifically designed to interrogate recurrent genetic variation with putative impact on gene function. We set out to test the hypothesis that variation within gene coding sequences is associated with CRC risk, by making use of the recently introduced Illumina Exon array.
Results
Post QC exome-wide analysis was based on 8,100 CRC cases and 21,820 controls from the six case-control series (Supplementary Tables 1 and 2). We also made use of genotypes for ~10,000 SNPs (~54% variants are non-synonymous) that were included in our previously published GWASs8,10, thus increasing power and providing additional exome array variant data on 4538 cases and 7225 controls (Supplementary Methods, Supplementary Table 3). Prior to the meta-analysis, we assessed the adequacy of the case-control matching and possibility of differential genotyping of cases and controls in individual studies using Quantile-Quantile (Q-Q) plots of test statistics (Supplementary Figure 6). Using data from the above 9 case-control series, we derived for each SNP joint odds ratios (ORs) and confidence intervals (CIs) in a meta-analysis under a fixed-effects model and determined the associated P values. Overall 72,162 non-monomorphic post-QC variants observed in at least 2 studies contributed to the combined meta-analysis totalling 12,638 cases and 29,046 controls (Supplementary Table 1). Of these variants, 29,117 variants were rare (MAF < 1%) and 32,809 variants exhibited MAF < 5%. We found no appreciable inflation of test statistics for the meta-analysis as a whole, λ90%bottom = 0.98, thereby excluding significant differential genotyping or cryptic population substructure (See Q-Q plot in Supplementary Figure 7)8,10,13.
Single variant analysis
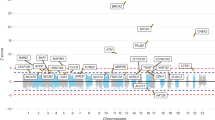
17 variants showed evidence for an association with CRC which exceeded Bonferroni-corrected exome-wide threshold of statistical significance (Table 1, Supplementary Table 4, Supplementary Figure 7), 4 of these 17 variants were non-synonymous missense variants: (rs3184504 (p.Trp263Arg) in SH2B3 (12q24; OR = 1.08, P = 3.9 × 10−7, effect allele frequency (EAF) = 0.52); rs16888728 (p.Pro215Gln) in UTP23 (8q24; OR = 1.15, P = 1.4 × 10−7, EAF = 0.10); two variants in FAM186A (12q13) - rs6580742 (p.Met2193Ile, OR = 1.11, P = 1.2 × 10−7, EAF = 0.19) and rs12303082 (p.Lys187Gln, OR = 1.09, P = 7.4 × 10−8, EAF = 0.36)). Another variant within 12q13 loci rs1129406 (12q13; OR = 1.11 P = 8.3 × 10−9, EAF = 0.41) is located within a splice region of ATF1. The rs3184504 association highlights a novel CRC risk locus (Table 1, Supplementary Figure 8). The p.Trp263Arg amino acid change resides in exon 3 of the SH2B adaptor protein and is predicted to be benign and tolerated by PolyPhen17 and SIFT18. Though predicted to be located within a transcription factor binding site (POLR2A) in lymphoblastoid, leukaemia and glioblastoma cell lines, it seems unlikely affect binding according to RegulomeDB (score 3a)19 or influence expression of SH2B3 in lymphoblastoid cell lines20,21 and other tissues22,23. Conditional analysis showed that rs3184504 genotype was sufficient to explain all of the effect at the 12q24 risk locus (Supplementary Table 5).
The 4 other novel SNPs rs16888728, rs6580742, rs12303082 and rs1129406 map to the previously described 8q24.1112,24 and 12q13.12 loci10 (Table 1). rs16888728 is located within exon 3 of UTP23 (8q23.3, 117783975, p.Pro215Gln) and is in moderate linkage disequilibrium (LD) with rs16892766 (8q23.3, 117630683)24 (D’ = 0.63, r2 = 0.30). Mutual adjustment was unable to distinguish the effects of rs16888728 on CRC risk from the previously described GWAS association, suggesting rs16892766 to be a primary signal (rs16888728, ORcond = 0.99, Pcond = 0.83; rs16892766, ORcond = 1.27, Pcond = 5.3 × 10−10) (Supplementary Table 6).
Detailed analysis of the 12q13 locus encompassing coding variants in ATF1 and FAM186A showed that three new variants are within a region of fairly extensive linkage disequilibrium (LD) (r2 = 0.31–0.68, D’ = 0.92–1) and in moderate LD with rs11169552, a previously identified through GWAS10 CRC risk locus (r2 = 0.08 − 0.24, D’ = 0.95–0.99). Both rs6580742 and rs12303082 are missense variants located within the exon 1 (rs6580742, chr12:50727811, p.Met2193Ile) and exon 3 (rs12303082, chr12:50754563, p.Lys187Gln) of FAM186A. Strongest signal at the locus (rs1129406) is a synonymous coding variant in ATF1 located within the splice region of gene, though it is unclear if the normal splicing of the gene is affected by the variant. rs6580742 is located within DNaseI hypersensitivity cluster and in eQTL with DIP2B and KIAA1463 expression in lymphoblastoid cell lines19,25,26 and cis-eQTL with ATF1 expression in esophagus mucosa, subcutaneous adipose tissue, tibial artery22,23. It is likely to affect binding according to RegulomeDB (score 1f)19,27. Conditional analyses indicate that all the association signals, including previously identified rs1116955210 (OR = 1.08, P = 2.55 × 10−5, ORcond = 1.02, Pcond = 0.35,EAF = 0.73), are explained by rs1129406, the splice region variant in ATF1 (Supplementary Table 7).
The remaining 10 SNPs in non-coding regions had been identified through our previous GWAS studies of CRC10,11,13,28,29,30. We subsequently applied conditional analysis to interrogate all CRC risk loci highlighted by the current study but found no evidence of multiple signals at 1q41, 8q24.21, 15q13.3, 18q21.1, 19q13.11, 20p12.3 and 20q13.33 (Supplementary Tables 8–14).
We further explored if rs1129406 (ATF1, 12q13), rs12303082 (FAM186A, 12q13), rs6580742 (FAM186A, 12q13), rs16888728 (UTP23, 8q24) and rs3184504 (SH2B3, 12q24) genotypes affect the CRC risk differentially by sex, age at diagnosis, tumor site, stage and MSI status (Supplementary Table 15). Intriguingly, we found that rs16888728 is significantly associated with gender in case-only analysis (OR = 1.21, P = 5.6 × −4) with no effect on CRC risk in males in case-control analysis (OR = 1.28, P = 5 × 10−8 in women and OR = 1.06 and P = 0.14 in men).
Gene-based analysis
Following on from these single variant analyses we conducted a gene-based analysis for rare (MAF < 1%) and low-frequency (MAF < 5%) variants observed in at least two cohorts (Supplementary Figure 9, Table 2). Meta-analysis of SKAT-O results showed some evidence of inflation (λ = 1.45 in analysis for low –frequency variants). Among the genes showing evidence of association in low-frequency variants analysis were tandemly located genes from protocadherin gamma gene cluster (PCDHGA3, PCDHGA2, PCDHGA1, PCDHGA4, PCDHGB1, 5q31.3, P < 2.9 × 10−6). The details of the SNPs contributing to PCDHG associations are given in Supplementary Table 16. None of the genes reached significance in rare – variant analysis.
Gene-ontology (GO) enrichment analysis implicated homophilic cell adhesion genes in CRC development (Supplementary Table 17).
Search for candidate high-penetrance CRC alleles
Next, we searched for rare high penetrance CRC variants by analysis of rare damaging variants present in more than 3 CRC cases, but absent from controls. In the analysis of dominant alleles, we observed truncating variants in NWD1, CD1A, ZNF594, DNAH9, ZNF418, ABTB1 and HIST1H3A and two missense variants in GCN1L1 (Supplementary Table 18). We also assessed the contribution of rare recessive alleles present in >3 cases, but absent in controls (Supplementary Table 18). Notable among these homozygotes were stop codon (p.Tyr90*) in the base excision repair gene, NTHL1, as well as homozygous missense variants in the DNA mismatch-excision repair gene, PMS1 (p.Thr75Ile) (Supplementary Figure 10). Overall we saw an excess of rare homozygous variants in base excision repair (16/8100 cases vs. 10/21820 controls, OR = 4.31; P = 2.4 × 10−4) and mismatch repair genes (11/8100 cases vs. 5/21820 controls, OR = 5.93, P = 6.1 × 10−4) in cases (Supplementary Table 19).
We also sought evidence of compound heterozygosity in cases and identified two damaging NOTCH2 variants and three damaging variants in DNAJC17 (DnaJ (Hsp40) homolog, subfamily C, member 17) that were observed to be present in heterozygous state at least twice in 2 and more cases, but absent in controls (Supplementary Table 20). NOTCH2 is regulated by Wnt signalling and known to have lower expression in colorectal and ovarian cancer31.
Discussion
We have identified coding variation in 4 genes (SH2B3, UTP23, FAM186A, ATF1) and PCDHG gene cluster that contribute to the risk of developing CRC. Three of the 4 genes with new coding variants influencing CRC risk had been identified by previous GWAS SNPs10,12,24. Novel association between the coding variant (rs3184504) in the SH2B3 gene has been described during the process of preparation and review of this manuscript in an independent meta-analysis32. Perhaps the most interesting finding of this well-powered study is the observation that very few recurrent coding sequence variants contribute to CRC risk and certainly not with major effect size (OR > 2.5).
The association between CRC risk and the adaptor protein, SH2B3, is interesting, since rs3184504 results in a predicted benign non-synonymous amino acid substitution (p.Trp263Arg) within the plekstrin homology domain of SH2B3. SH2B3 is induced upon JAK-STAT3 phosphorylation and is expressed at high levels in haematopoietic cells, but only at low levels in the normal colon. The protein is a regulator of cytokine signals at the cell surface through tyrosine kinase signalling cascades and is thought to act as a negative regulator of such signals at the cell surface to impart an anti-proliferative effect. A consanguineous family has been reported which segregates a germline frameshift mutation in the Plekstrin homology domain of SH2B3. Homozygous individuals developed various autoimmune phenotypes and one sibling developed acute lymphoblastic leukaemia (ALL) as an infant33. Somatic SH2B3 mutations have also been identified in 3% of ALL, suggesting that SH2B3 loss plays a role in initiation and progression of human leukaemia through dysregulated cytokine signalling. Interrogation of TCGA and Broad Institute sequence data from colorectal adenocarcinomas34,35,36 did not identify an excess of somatic mutations in SH2B3 (0.69% of samples carry deleterious mutations or copy number variations), suggesting that SH2B3 mutations are not drivers in CRC progression33. Genetic variation at the SH2B3 gene locus has been associated with various autoimmune related disorders including hepatitis37, rheumatoid arthritis38, hypothyroidism39, type 1 diabetes40, vitiligo41, rheumatoid arthritis and coeliac syndrome42, suggesting that SH2B3 dysfunction may be involved in mediating disordered immune function and thereby play a role in cancer susceptibility. Interestingly, SH2B3 is over-expressed in ovarian tumour cells with evidence for a role in activating signal transduction43. SH2B3 expression status may have paradoxical effects in cancer, dependent on cellular context.
The variant in UTP23 (rs16888728) also exerts a modest effect on CRC risk. The UTP23 transcript is expressed at modest levels in many tissue types. It has sequence homology to a yeast protein involved in ribosomal RNA processing and ribosome biogenesis. As such, it may be involved in alternative splicing, although very little is known about the functional role of the human protein. The coding variant (rs16888728) is located within exon 3 of UTP23 and results in a non-conserved amino acid substitution (p.Pro215Gln, GERP score = −0.543). Conditional analysis was unable to distinguish the effects of rs16888728 on CRC risk from that of the previously described24 GWAS association (rs16892766). Interrogation of tumour sequence databases reveals no significant excess of mutations in CRC (<1% prevalence)34,35,36. However, UTP23 is amplified in ~5% of CRC tumours35,36 with significant correlation between UTP1 mRNA expression and copy number variation.
The SNP rs1129406, a splice site variant in ATF1, appears to explain the association signal at the 12q13 locus, including that of a previous signal identified by GWAS (rs11169552)10. ATF1 is a transcription factor that, when phosphorylated, induces transcriptional transactivation of target genes. Fusion of ATF1 with the Ewing’s Sarcoma gene, or with FUS, results in continuous signaling and sarcomatous tumour formation. Common variation has not been associated with other cancers, however significant cis-eQTL with ATF1 was detected for this variant in esophagus mucosa, subcutaneous adipose tissue and tibial artery22,23. Whilst there are no excess of somatic mutations in CRC tissue in TCGA or Broad data, rs1129406 may be the causative variant that explains the previous GWAS signal. The relationship of FAM186A to CRC risk is somewhat opaque, as very little is known about this gene. FAM186A appears to be a protein coding gene, rather than a lncRNA. Hence we cannot exclude the possibility that the effect is mediated through regulatory effects.
The gene-based test, SKAT-O, highlighted several genes from protocadherin gamma (PCDHG) gene cluster on chromosome 5 exhibiting a composite excess of coding variants and thereby indicating the gene is associated with CRC risk. Somatic genomic missense and nonsense mutations in one of the identified genes are present in 11.8% of CRC cases and up to 31% of all skin cuteneous melanomas (according to The Cancer Genome Atlas data)35. PCDHG gene cluster encodes 22 genes divided into 3 subfamily (A,B and C) based on sequence similarities with multiple transcripts generated by alternative splicing44. PCDH expression is observed in colon and long range epigenetic silencing of PCDH cluster region has been described in Wilm’s tumours45, breast cancers46 and colorectal adenomas and carcinomas47. Hence, PCDH genes play role of tumour suppressor and silencing mutations might be expected to have tumour-promoting effects. Whilst PCDHG cluster genes are strong candidates based on the analysis presented in this study, further work is required to confirm the role of these genes in cancer predisposition.
The identification of damaging alleles acting as rare recessive traits in genes that participate in DNA repair, with known paradigms in CRC susceptibility, such as NTHL1 (p.Tyr90*) and PMS1 (p.Thr75Ile) clearly require further study as these represent strong candidate recessive alleles. Recently NTHL1 loss-of-function germline mutation has been described in families with adenomatous polyposis and progression to CRC inherited in recessive mode48, thus suggesting that the observed association is real and our search for rare damaging alleles is a successful approach to identify candidate variants. The observed excess of rare damaging variants in base-excision and mismatch repair genes suggests that the clinical importance of moderately penetrant, disease-causing, variants in DNA repair genes may be underestimated. However, further studies will require even larger sample sizes, given the rarity of the alleles, unless sequencing can identify new alleles in addition to those catalogued here. Indeed, many of the genes with damaging variants represent strong candidates for validation in exome and whole genome sequencing efforts.
Given the expectation that uncommon functional variation might be associated with CRC risk, with larger effect size than common variation, it is surprising that we have identified so few new coding sequence variants and that all of these exert modest effect sizes (OR 1.08–1.15). In a linear-mixed model analysis (Supplementary Material), we estimated that the genetic variants identified though previous GWAS and significant in our meta-analysis explain approximately 1.5 ± 0.7% of the total phenotypic variance on the liability scale, while the newly identified variants account for only 0.4% of the total variance.
The Infinium Human Exome BeadChip 12v1.0 or 12v1.1 (Illumina Inc.) array was configured to identify coding sequence variants most likely to have functional consequences. Despite of its attractiveness as a cheap alternative to exome sequencing, exome array has some limitations and is not able to offer complete whole exome coverage of all possible functional variants and indels. Importantly, exome array was designed based on exome sequencing of 12,000 samples and enriched for multiple outcomes such as cardiovascular disease, obesity, diabetes, autism and cancer49, which may not be representative of our cohorts. There were some differences in the genotyping quality between various versions of arrays used in the analyses and many variants did not pass stringent quality control criteria. Around 70,000 SNPs were non-monomorphic in European populations, present in at least two studies and passed our QC measures.
The focus on genetic variants with potential detrimental functional consequences should also enhance the a priori likelihood of pathogenicity. Though limited in detection of indels with only 136 present on the chip, the study was well powered to detect plausible effect sizes and allele frequencies (Supplementary Figure 11). Indeed, the study size had 80% power to detect an OR > 3 provided the MAF was >0.001 and an OR odds >1.8 if the MAF was 0.005. Whilst larger studies and/or meta-analysis might identify further coding variants with functional effects, the paucity of findings of recurrent low frequency coding variation impacting on CRC risk is intriguing. Because the causative gene mutations have been characterised for almost all dominant high penetrance CRC families, it seems unlikely that rare recurrent alleles in European populations have yet to be identified with large effects (OR > 5), apart from private mutations or recessive traits that are unlikely to be discovered through designed commercial arrays. Hence, population-specific custom exome arrays as well exome and genome sequencing of trios and families may be a way forward to identify recurrent rare genetic variation of moderate effect of risk and private mutations.
Materials and Methods
Study populations
The study was based on six independent case control series from European populations including Scotland (3,616 cases and 10,312 controls), England (4,558 cases and 11,249 controls), Germany (284 cases and 1,100 controls), Holland (480 cases and 480 controls), Spain (300 cases and 300 controls) and Portugal (200 cases and 200 controls). Details regarding these participating studies are described in the Supplementary Data (available online). All cases had histologically confirmed adenocarcinoma of the colon or rectum (codes 153 or 154 International Classification of Diseases (ICD), 9th revision or ICD10 C18, C19 or C20 codes). The study was undertaken at participating centres with written informed consent in accordance with respective Institutional Review Boards (IRB)/Ethics Committees.
To enhance our power we made use of previously published GWASs8,10 thus providing ~10.000 exome array variant data on 3,549 cases and 3,698 controls from UK1 and UK2 studies, 3,158 cases and 3,073 controls from Scotland Phase1, Scotland Phase2 and Scotland Phase3 and 1,794 cases and 2,686 controls from the VQ58 study8,13 (Supplementary Methods, Supplementary Tables 2, 3). After quality control and exclusion of expected and unexpected duplicates between studies we ended up with exome array variant data on 3,033 cases and 3,690 controls from UK1 and UK2 studies, 556 cases and 2,997 controls from Scotland Phase1, Scotland Phase2 and Scotland Phase3 and 949 cases and 538 controls from the VQ58 study8,13. Study details, details of genotyping, quality control procedures, sample and SNPs exclusion for these GWAS-focussed studies have been published previously8 (Supplementary Data, Supplementary Tables 2, 3).
Exome Array Genotyping and Quality Control
DNA was extracted from EDTA-venous blood samples using standard methodologies at each centre. Genotyping was performed using the Infinium Human Exome BeadChip 12v1.0 or 12v1.1 (Illumina Inc., San Diego, CA), with genotype calling using Illumina GenCall for HumanExome-12v1.0 and HumanExome-12v1.1 versions called separately. Generation Scotland controls and a subset of the cases from the SOCCS study were genotyped using OmniExpressExome BeadChip 8v1.1 or 8v1.250 (Illumina Inc., San Diego, CA). A summary of the array SNP content51,52 and the respective SNP inventory53 have been provided previously. Standard quality procedure were applied, with further details of sample and probe exclusion in Supplementary Material and Supplementary Table 2. We compared MAF and genotyping call genotyping call rates between different version of arrays used in the current study and excluded all variants that showed some evidence of differences (Supplementary Figures 1,3). Additionally, we compared allele frequency to the 1000G data and UK exome array consortium (Supplementary Figure 2). Following standard quality-assurance and quality control measures this collaborative initiative provided information on 12,638 CRCs cases and 29,045 controls (Supplementary Table 1).
Statistical analysis
We designed the study according to an estimate of the sample size required to detect plausible effect sizes (OR = 1.5–5.0) at various rare allele frequencies (>0.001). Following completion of the study and all QC measures, we re-estimated statistical power for a given sample size using QUANTO version 1.2.454 for the main effect of genetic variant and the log-additive model of inheritance stipulating a P-value of 5.5 × 10−7, which corresponds to Bonferroni-corrected exome-wide level of significance.
The association between individual variants and risk of CRC was evaluated in initial data analysis using unconditional logistic regression under a log-additive model of inheritance for each study separately. To examine whether associations at each identified locus were independent, we conducted conditional analysis by controlling for allelic dosage for the most significantly associated SNP at the locus. We subsequently applied conditional analysis to interrogate following CRC risk loci highlighted by the current study: 1q41 controlling for rs6687758, 8q23.3 controlling for rs16892766 and/or rs16888728, 8q24.21 controlling for rs10505477, rs6983267 and/or rs7014346, 11q32.1 controlling for rs3802842, 12q13.12 controlling for rs6580742, rs12303082 and rs1129406, 12q24.12 controlling for rs3184504, 14q22.2 controlling for rs4444235, 15q13.3 controlling for rs4779584, 18q21.1 controlling for rs4939827, 19q13.11 controlling for rs10411210, 20p12.3 controlling for rs961253 and 20q13.33 controlling for rs4925386.
Individual study effect estimates (Odds ratios (OR) and associated 95% confidence intervals (CIs)) derived from logistic regression were combined in a meta-analysis. We used a fixed effect inverse variance weighting model for meta-analysis to maximize discovery power of the current study55. Only non-monomorphic variants observed in at least two studies were included in the meta-analysis. We tested for over-dispersion of P-values in the meta-analysis by generating quantile-quantile (QQ) plots and deriving an inflation factor (λ). Cochran’s Q statistic was used to test for heterogeneity and the I2 statistic to quantify the proportion of the total variation due to heterogeneity. I2 values ≥ 75% were considered to indicate excessive heterogeneity56 and variants displaying I2 values > 75% in were excluded from further analysis. Taking all the above measures into account, 72,162 SNPs remained in the analysis, equating to a Bonferroni-corrected exome-wide threshold of statistical significance of 5.55 × 10−7. This is conservative given the likely linkage disequilibrium between some variants. We further examined top variants and excluded those that showed obvious problems with clustering and differences in clustering between versions of genotyping platforms in our analysis. This included monomorphic rs1058065 (exm2255298).
Association by sex, age, stage (invasive, non-invasive), MSI status and tumour site (rectal [ICD9:154], colonic [ICD9:153]) for the top new variants were further explored using ordered logistic regression in case-only analysis. All statistical tests were two-sided.
Gene based and pathway analysis
To explore the effects of more than one variant in the same gene on CRC risk, we used the small-sample-adjusted unified test, SKAT-O57 with default weight on rare variants. All variants observed in at least two studies contributed to the SKAT-O results. We performed analyses for rare (MAF > 1%) and low frequency variants (MAF below 5%) including all and only High and Moderate effects as annotated by SnpEff 58. Due to the different number of variants in each individual study we performed SKAT-O test separately for each individual study and combined summary statistics from individual SKAT results in a meta-analysis using “MetaSKAT” package in R59 Similarly to single-variant analysis we tested for over-dispersion of P-values by generating QQ plots and deriving an inflation factor (λ). To account for multiple testing in these gene-based tests, we set the significance threshold to be P < 2 × 10−6 to reflect Bonferroni correction for the 23,280 genes examined. These 23,280 genes were selected on the base of the presence of 2 and more variants per gene and unique mapping coordinates. We further examined top genes and excluded those that were driven by single variant with the differences in clustering between versions of genotyping platforms in our analysis. This included monomorphic rs1058065 (EIF2B4) .
Further, we investigated variants contributing to the gene-based test. To determine whether genes identified in SKAT-O were enriched for particular molecular pathways, we performed a gene ontology (GO) enrichment analysis on a sorted by p value list of genes , using Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla)60,61.
Search for candidate high-penetrance CRC alleles
We considered the possibility that rare damaging variants represented on the exome array might confer high-penetrance susceptibility to CRC and conducted exploratory data analysis. We reasoned on the basis of pre-existing empiric data that any dominant alleles would be likely to have frequencies of <0.1%, whereas recessive alleles would have frequencies of <2% in controls. Dominant alleles were filtered from the entire variant set as follows: [1] predicted not to be benign/tolerated by both SIFT18 and PolyPhen217 or nonsense variants; [2] excluded probable miscalled SNPs through visual inspection of genotyping clusters; [3] absent in controls to ensure inclusion of potentially high penetrance risk alleles. Recessive alleles were filtered from the entire variant set as follows: [1] predicted not benign or tolerated by both SIFT18 and PolyPhen217; [2] excluded probable miscalled SNPs through visual inspection of genotyping; [3] homozygotes absent in controls to ensure inclusion of potentially high penetrance risk alleles; [4] minor allele frequency ≤ 0.02 in controls.
We evaluated effect of rare damaging variants under dominant or recessive model of inheritance using Fisher’s exact test in a pooled analysis. Due to the limited number of rare damaging variants on traditional GWAS platforms, we included in the analysis case-control series genotyped using Exome Array only (8100 cases/21820 controls). We also looked for evidence of an excess of compound heterozygosity for rare damaging variants in cases compared to controls. The compound heterozygous list was filtered from the entire set of heterozygous variants as follows: (1) excluded probable miscalled SNPs through visual inspection of genotyping clusters, [2] predicted not to be benign/tolerated by both SIFT18 and PolyPhen217, (3) number of rare damaging heterozygotes per gene in controls ≤ 1, (4) minor allele frequency ≤ 2% in controls. We further look for excess of rare damaging homozygous variants in DNA repair pathways by counting number of homozygous rare variants in cases and controls and testing significance by Fisher exact test.
Although this study did not have power to detect such alleles by association testing or by gene burden tests, we catalogued all candidate alleles that fulfilled these criteria.
Additional Information
How to cite this article: Timofeeva, M. N. et al.Recurrent Coding Sequence Variation Explains Only A Small Fraction of the Genetic Architecture of Colorectal Cancer. Sci. Rep.5, 16286; doi: 10.1038/srep16286 (2015).
References
Lichtenstein, P. et al.Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark and Finland. N Engl J Med 343, 78–85, doi: 10.1056/NEJM200007133430201 (2000).
Czene, K., Lichtenstein, P. & Hemminki, K. Environmental and heritable causes of cancer among 9.6 million individuals in the Swedish Family-Cancer Database. Int J Cancer 99, 260–266, doi: 10.1002/ijc.10332 (2002).
Jiao, S. et al.Estimating the heritability of colorectal cancer. Hum Mol Genet 23, 3898–3905, doi: 10.1093/hmg/ddu087 (2014).
Wang, H. et al.Trans-ethnic genome-wide association study of colorectal cancer identifies a new susceptibility locus in VTI1A. Nat Commun 5, 4613, doi: 10.1038/ncomms5613 (2014).
Zhang, B. et al.Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 46, 533–542, doi: 10.1038/ng.2985 (2014).
Whiffin, N. et al.Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet, doi: 10.1093/hmg/ddu177 (2014).
Peters, U. et al.Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology 144, 799–807 e724, doi: 10.1053/j.gastro.2012.12.020 (2013).
Dunlop, M. G. et al.Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet 44, 770–776, doi: 10.1038/ng.2293 (2012).
Kinnersley, B. et al.The TERT variant rs2736100 is associated with colorectal cancer risk. Br J Cancer 107, 1001–1008, doi: 10.1038/bjc.2012.329 (2012).
Houlston, R. S. et al.Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 42, 973–977, doi: 10.1038/ng.670 (2010).
Houlston, R. S. et al.Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 40, 1426–1435, doi: 10.1038/ng.262 (2008).
Peters, U. et al.Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet 131, 217–234, doi: 10.1007/s00439-011-1055-0 (2012).
Tenesa, A. et al.Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40, 631–637, doi: 10.1038/ng.133 (2008).
Welter, D. et al.The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–1006, doi: 10.1093/nar/gkt1229 (2014).
Tomlinson, I. P. et al.Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4 and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet 7, e1002105, doi: 10.1371/journal.pgen.1002105 (2011).
McClellan, J. & King, M. C. Genetic heterogeneity in human disease. Cell 141, 210–217, doi: 10.1016/j.cell.2010.03.032 (2010).
Adzhubei, I. A. et al.A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi: 10.1038/nmeth0410-248 (2010).
Ng, P. C. & Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res 11, 863–874, doi: 10.1101/gr.176601 (2001).
Boyle, A. P. et al.Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797, doi: 10.1101/gr.137323.112 (2012).
Stranger, B. E. et al.Patterns of cis regulatory variation in diverse human populations. PLoS Genet 8, e1002639, doi: 10.1371/journal.pgen.1002639 (2012).
Yang, T. P. et al.Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 26, 2474–2476, doi: 10.1093/bioinformatics/btq452 (2010).
The GTEx Consortium . Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660, doi: 10.1126/science.1262110 (2015).
The GTEx Consortium . (2015) Available at: http://www.gtexportal.org (Accessed: 19 August 2015).
Tomlinson, I. P. et al.A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 40, 623–630, doi: 10.1038/ng.111 (2008).
Stranger, B. E. et al.Population genomics of human gene expression. Nat Genet 39, 1217–1224, doi: 10.1038/ng2142 (2007).
Veyrieras, J. B. et al.High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4, e1000214, doi: 10.13JU71/journal.pgen.1000214 (2008).
RegulomeDB . Available at: http://regulomedb.org/ (Accessed: December 2014).
Broderick, P. et al.A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 39, 1315–1317, doi: 10.1038/ng.2007.18 (2007).
Tomlinson, I. et al.A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39, 984–988, doi: 10.1038/ng2085 (2007).
Zanke, B. W. et al.Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39, 989–994, doi: 10.1038/ng2089 (2007).
Chu, D. et al.Notch2 expression is decreased in colorectal cancer and related to tumor differentiation status. Ann Surg Oncol 16, 3259–3266, doi: 10.1245/s10434-009-0655-6 (2009).
Schumacher, F. R. et al.Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun 6, 7138, doi: 10.1038/ncomms8138 (2015).
Perez-Garcia, A. et al.Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood 122, 2425–2432, doi: 10.1182/blood-2013-05-500850 (2013).
Lawrence, M. S. et al.Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501, doi: 10.1038/nature12912 (2014).
Gao, J. et al.Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1, doi: 10.1126/scisignal.2004088 (2013).
Cerami, E. et al.The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404, doi: 10.1158/2159-8290.CD-12-0095 (2012).
de Boer, Y. S. et al.Genome-wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology 147, 443–452 e445, doi: 10.1053/j.gastro.2014.04.022 (2014).
Okada, Y. et al.Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381, doi: 10.1038/nature12873 (2014).
Eriksson, N. et al.Novel associations for hypothyroidism include known autoimmune risk loci. PLoS One 7, e34442, doi: 10.1371/journal.pone.0034442 (2012).
Plagnol, V. et al.Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS Genet 7, e1002216, doi: 10.1371/journal.pgen.1002216 (2011).
Jin, Y. et al.Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet 44, 676–680, doi: 10.1038/ng.2272 (2012).
Zhernakova, A. et al.Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet 7, e1002004, doi: 10.1371/journal.pgen.1002004 (2011).
Ding, L. W. et al.LNK (SH2B3): paradoxical effects in ovarian cancer. Oncogene, doi: 10.1038/onc.2014.34 (2014).
Morishita, H. & Yagi, T. Protocadherin family: diversity, structure and function. Curr Opin Cell Biol 19, 584–592, doi: 10.1016/j.ceb.2007.09.006 (2007).
Dallosso, A. R. et al.Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet 5, e1000745, doi: 10.1371/journal.pgen.1000745 (2009).
Novak, P. et al.Agglomerative epigenetic aberrations are a common event in human breast cancer. Cancer Res 68, 8616–8625, doi: 10.1158/0008-5472.CAN-08-1419 (2008).
Dallosso, A. R. et al.Long-range epigenetic silencing of chromosome 5q31 protocadherins is involved in early and late stages of colorectal tumorigenesis through modulation of oncogenic pathways. Oncogene 31, 4409–4419, doi: 10.1038/onc.2011.609 (2012).
Weren, R. D. et al.A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature genetics 47, 668–671, doi: 10.1038/ng.3287 (2015).
Lee, S., Abecasis, G. R., Boehnke, M. & Lin, X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 95, 5–23, doi: 10.1016/j.ajhg.2014.06.009 (2014).
Illumina . Infinium OmniExpressExome-8 BeadChip. Available at: http://support.illumina.com/array/array_kits/infinium_humanomniexpressexome_beadchip_kit.html (Accessed: 23 April 2014).
Illumina . datasheet_humanexome_beadchips.pdf Available at: http://products.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_humanexome_beadchips.pdf (Accessed: 27 April 2014).
Exome Chip Design . (2013) Available at: http://genome.sph.umich.edu/wiki/Exome_Chip_Design (Accessed: 1 September 2014).
Illumina . HumanExome-12v1-2_A.annotated.txt. Available at: ftp://webdata2:webdata2@ussd-ftp.illumina.com/downloads/ProductFiles/HumanExome-12/HumanExome-12v1-2_A.annotated.txt (Accessed: 30 September 2014).
Gauderman, W. J. Sample size requirements for matched case-control studies of gene-environment interaction. Stat Med 21, 35–50 (2002).
Pereira, T. V., Patsopoulos, N. A., Salanti, G. & Ioannidis, J. P. Discovery properties of genome-wide association signals from cumulatively combined data sets. Am J Epidemiol 170, 1197–1206, doi: 10.1093/aje/kwp262 (2009).
Higgins, J. P. & Thompson, S. G. Quantifying heterogeneity in a meta-analysis. Stat Med 21, 1539–1558, doi: 10.1002/sim.1186 (2002).
Lee, S. et al.Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 91, 224–237, doi: 10.1016/j.ajhg.2012.06.007 (2012).
Cingolani, P. et al.A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92, doi: 10.4161/fly.19695 (2012).
Lee, S., Teslovich, T. M., Boehnke, M. & Lin, X. General framework for meta-analysis of rare variants in sequencing association studies. Am J Hum Genet 93, 42–53, doi: 10.1016/j.ajhg.2013.05.010 (2013).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48, doi: 10.1186/1471-2105-10-48 (2009).
Eden, E., Lipson, D., Yogev, S. & Yakhini, Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol 3, e39, doi: 10.1371/journal.pcbi.0030039 (2007).
Acknowledgements
We thank the participants in all of the studies that contributed to this piece of work and all the recruitment teams and collaborators who make such studies possible. Generation Scotland is a collaboration between the University Medical Schools and NHS in Aberdeen, Dundee, Edinburgh and Glasgow. We are grateful to GPs and Scottish School of Primary Care for help with recruitment to GS:SFHS. We align="center"owledge the expert technical support on sample preparation and genotyping by the Genetics Core of the Edinburgh University Wellcome Trust Clinical Research Facility. We are grateful to many colleagues within UK Clinical Genetics Departments (CORGI) and to many collaborators who participated in the VICTOR and QUASAR2 trials. We also thank colleagues from the UK National Cancer Research Network (for NSCCG). The study made use of genotyping data from the 1958 Birth Cohort, kindly made available by the Wellcome Trust Case Control Consortium 2. A full list of the investigators who contributed to the generation of the data is available at http://www.wtccc.org.uk/. We are grateful to all members of UK exome array consortium for sharing information on allele frequencies (http://diagram-consortium.org/uk-exome-chip/). We also want to align="center"owledge computer and cluster service at IGMM and Martin Taylor for an access to his group computer facilities. Funding: In Scotland, the work was funded by a Cancer Research UK Programme Grant (C348/A12076) (MGD, MT, SMF, VS, LYO, LZ, ET, AT, HC) and Medical Research Council Grant (MR/KO18647/1) (SMF, MGD). The Generation Scotland Family Health Study (DP, AC, CH) was funded by a grant from the Scottish Government Health Department (CZD/16/6). Support to the Lothian Birth Cohort (IJD, SEH): genotyping by Biotechnology and Biological Sciences Research Council (BBSRC); phenotype collection (LBC 1921) by BBSRC, The Royal Society and The Chief Scientist Office of the Scottish Government; phenotype collection for the LBC 1936 by Age UK-The Disconnected Mind project. Funding also from the cross council Lifelong Health and Wellbeing Initiative (G0700704/84698). At the Institute for Cancer Research (RSH, BK, NW, AL, FH, SD, PB) the work was funded by a Cancer Research UK Programme Grant from the Bobby Moore Fund (C1298/A8362). In Leeds (DTB), the work was funded by Cancer Research UK Programme Grant (C588/A19167). BK is supported by a PhD studentship funded by the Sir John Fisher Foundation. In Oxford (IPMT, CP, MG, EB, LM), the work was funded by: CTUK programme grant to IPMT; Oxford Comprehensive Biomedical Research Centre (CP, IPT); EU FP7 CHIBCHA grant (IPMT); core infrastructure support to the Wellcome Trust Centre for Human Genetics, Oxford (Wellcome Trust 090532/Z/09/Z). In the Netherlands (HM, DR, CT, JW, MS, AB, HFAV, FJH, TvW) funding was from the Association for International Cancer Research (10-0619) and the Dutch Cancer Society (UL2010-4656). In Germany (AF, WL, CS, JH, SB) funding was as follows: POPGEN 2.0 network is supported by a grant from the German Ministry for Education and Research (01EY1103). Kiel: The study was supported by the German National Genome Research Network (NGFN) through the POPGEN biobank (BmBF 01GR0468) and the National Genotyping Platform. The studies in Portugal (MP CP MT) were funded by CHIBCHA and Liga Portuguesa Contra o Cancro. In Spain (CR-P, AC, SC-B, AC) the work was supported by grants from the Fondo de Investigación Sanitaria/FEDER (11/00219, 11/00681, PI13/01136), Ministerio de Economía y Competitividad (SAF2010-19273), Fundación Privada Olga Torres, Fundación Científica de la Asociación Española contra el Cáncer (GCB13131592CAST), Asociación Española de Gastroenterología (beca grupo de trabajo “Oncología”, Agència de Gestió d’Ajuts Universitaris i de Recerca (Generalitat de Catalunya,2009SGR849, 2014SGR135, 2014SGR255), FP7 CHIBCHA Consortium and by COST office through COST action BM1206. SCB is supported by the Fondo de Investigación Sanitaria (CP 03-0070). In addition, this work also received support from the COST Action BM1206 (all groups). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
M.N.T., B.K., I.P.M.T., M.G.D. and R.S.H. contributed to writing of the manuscript. M.N.T., B.K., S.M.F., H.C., D.T.B., I.P.M.T., M.G.D. and R.S.H. conceived and designed the experiments. M.N.T., B.K., S.M.F., L.Y.O., I.P.M.T., M.G. and R.S.H. performed the experiments. M.N.T., B.K., V.S., L.Y.O., G.G., I.P.M.T., M.G.D. and R.S.H. analysed the data. M.N.T., B.K., S.M.F., N.W., C.P., V.S., A.L., M.G., L.Y.O., F.H., E.B., L.Z., S.D., L.M., E.T., P.B., A.T., G.G., C.H., A.C., I.J.D., S.E.H., E.N., J.B., G.S., R.W., D.F., H.M., D.R., C.T., J.W., M.S., A.B., H.F.A.V., F.J.H., T.W., A.F., W.L., C.S., J.H., S.B., P.P., K.H., A.F., H.W., R.H., M.P., C.P., M.T., C.R.-P., A.C., S.C.-B., A.C., H.C., D.T.B., I.P.M.T., M.G.D. and R.S.H. were involved in study design/sampling/ assembly/data collection, collation, curation and qulaity control/data analysis from case-control cohorts for respective centres. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Timofeeva, M., Kinnersley, B., Farrington, S. et al. Recurrent Coding Sequence Variation Explains Only A Small Fraction of the Genetic Architecture of Colorectal Cancer. Sci Rep 5, 16286 (2015). https://doi.org/10.1038/srep16286
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16286
This article is cited by
-
Novel significant stage-specific differentially expressed genes in hepatocellular carcinoma
BMC Cancer (2019)
-
Genome-wide scan of the effect of common nsSNPs on colorectal cancer survival outcome
British Journal of Cancer (2018)
-
Exploring causality in the association between circulating 25-hydroxyvitamin D and colorectal cancer risk: a large Mendelian randomisation study
BMC Medicine (2018)
-
Evidence for genetic association between chromosome 1q loci and predisposition to colorectal neoplasia
British Journal of Cancer (2017)
-
Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer
Nature Communications (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
