
Abstract
Bacterial pathogens subvert signalling pathways to promote invasion and/or replication into the host. LegK1-4 proteins are eukaryotic-like serine/threonine kinases that are translocated by the Dot/Icm type IV secretion system (T4SS) of several Legionella pneumophila strains. We present the crystal structures of an active fragment of the LegK4 protein in apo and substrate-bound states. The structure of LegK41–445 reveals a eukaryotic-like kinase domain flanked by a novel cap domain and a four-helix bundle. The protein self-assembles through interactions mediated by helices αF and αG that generate a dimeric interface not previously observed in a protein kinase. The helix αG is displaced compared to previous kinase structures and its role in stabilization of the activation loop is taken on by the dimerisation interface. The apo-form of the protein has an open conformation with a disordered P-loop but a structured activation segment in absence of targeted phosphorylation. The nucleotide-binding site of LegK4 contains an unusual set of residues that mediate non-canonical interactions with AMP-PNP. Nucleotide binding results in limited changes in the active site, suggesting that LegK4 constitutive kinase activity does not depend on phosphorylation of the activation loop but on the stabilizing effects of the dimer.
Similar content being viewed by others


Introduction
Protein phosphorylation is one of the most common mechanisms of regulation and signalling1. If protein phosphorylation on Ser, Thr and Tyr residues was for a long time perceived as a eukaryote-specific activity, it is now a well-established signalling mechanism in bacteria2 and archaea3. Eukaryotic kinases are grouped together in the eukaryotic protein kinase superfamily based on sequence homology between their kinase domains4. These domains are typically organized into 12 conserved subdomains that fold in a characteristic two-lobed catalytic core structure, with the catalytic active site lying in a deep cleft formed between the two lobes (reviewed in5). The N-terminal lobe is involved primarily in binding and orienting the phospho-donor ATP molecule, whereas the C-terminal lobe binds the protein substrate and initiates the transfer of the phosphate group. The structural basis governing kinase activation has been widely documented (see review6). While kinases adopt a variety of conformations in their inactive states, active kinases adopt a single arrangement of conserved structural features. This includes two sets of hydrophobic residues that form spines running through the interior of the kinase which when properly formed, secure the proper positions of catalytic residues7. The C-spine contributes to the binding of ATP and is completed by the adenine base. Formation of the R-spine is coupled to the stabilization of the activation loop, a region that contains one or more sites of activating phosphorylation within most kinases.
Because of its essential role in eukaryotic cells, interfering with host-cell proteins phosphorylation is a common strategy used by bacterial pathogens to hijack cell functions8. A growing number of bacterial effectors targeting phosphorylation pathways have been shown to play a role in infection (reviewed in9). Among them, a number of effector kinases are found to be injected into host-cell via dedicated machinery including type 3 (T3SS) or type 4 (T4SS) secretion systems. Recent structural studies have shed light onto some of these bacterial effectors kinases10. These include NleH1 and NleH2 from entheropathogenic and enterohaemorrhagic Escherichia coli11, CtkA from Helicobacter pylori12, OspG from Shigella13,14. Although these proteins belong to the kinase family, they were found to be clearly divergent from a sequence and structural point of view with eukaryotic kinases. In particular, only the motifs I to VII of characteristic 12 motifs are present and these effectors lack an activation loop as well as helices αG, αH and αI (also named ‘GHI domain’ in10).
Other effector kinases seem to have been acquired through gene transfer and can be considered as eukaryotic-like kinases. These include Salmonella Typhimurium SteC, Yersinia pestis YpkA and Legionella pneumophila LegK kinases family. The later stands out as unique given that four of these proteins are translocated via the Dot/Icm T4SS15. L. pneumophila is an intracellular Gram-negative bacterium able to infect human alveolar macrophages and to cause Legionnaires’ disease, a severe pneumonia. Instrumental to its survival in infected cells is L. pneumophila capacity to re-route the host vesicle trafficking pathways so that it avoids fusion with lysosomes and safely multiplies inside ER-like membrane bound vacuoles (Legionella containing vacuoles or LCVs)16. The Dot/Icm T4SS plays a key role in infection and is absolutely required for the intracellular multiplication of the pathogen17,18. The Dot/Icm system translocates nearly 300 effectors (reviewed in19,20,21), which target various cellular pathways such as anti-apoptosis22 and vesicle trafficking23, to modify the cell environment in favour of L. pneumophila replication.
Sequence analysis of LegK proteins showed that they range from 50 to 100 kDa in mass and share a eukaryotic-like Ser/Thr kinase domain containing the Hanks motifs15. While belonging to the same family of effectors, the functions of the LegK proteins are not redundant. On the one hand, LegK1 indirectly regulates the expression of a group of host genes involved in inflammation and anti-apoptosis by inducing the specific activation of the innate immunity pathway NF-κB24. The same study demonstrated that LegK1 mimics IκB kinase (IKK) by directly phosphorylating a group of NF-κB inhibitors belonging to the IκB family. On the other hand, LegK2 was found to target a different pathway. Inactivation of LegK2 causes a significant decrease in the virulence of L. pneumophila and a delay in the intracellular replication compared to the wild strain15. More precisely, LegK2 has been recently shown to phosphorylate the host actin nucleator complex Arp2/3, thus inhibiting actin polymerisation on the LCV and consequently the late endosomes trafficking towards the phagosome. Thus, LegK2 directly contributes to Legionella evasion from the endocytic degradation25. Little is known about the functions of LegK3 and LegK4. LegK4 is the largest protein of the family with a large C-terminal domain in addition to the putatively conserved N-terminal functional region containing the kinase domain.
To gain insight into this novel family of T4SS effectors, we have solved the crystal structure of a catalytically active fragment of LegK4 (residues 1–445, LegK41–445) in apo and AMP-PNP bound states. These structures reveal a eukaryotic-like kinase motif and two previously uncharacterized domains. LegK41–445 kinase domain displays unique features, including a modified nucleotide-binding pocket and an original position of the αG helix. A novel mode of dimerisation of the kinase domain was identified in the crystal structures and confirmed by small angle X-ray scattering (SAXS). Our study demonstrates that LegK41–445 is constitutively active and suggests that the dimer assembly stabilizes the active conformation.
Results and Discussion
LegK41–445 is active for auto- and substrate phosphorylation
In the process of trying to crystallise LegK4 we observed partial degradation of the full-length protein during purification and identified a stable fragment encompassing residues 1 to 445 (LegK41–445). This fragment corresponds to the most conserved region in the LegK family15. We thus generated an expression vector corresponding to this fragment (Fig. 1a) and purified the protein. To determine if LegK41–445 was active, a phosphorylation assay was conducted with either the full length LegK4 or the LegK41–445 fragment. The results show that, as for the wild type, LegK41–445 was able to autophosphorylate using [γ-32P]ATP as substrate (Fig. 1b). Moreover, both wild type LegK4 and LegK41–445 were able to phosphorylate the myelin basic protein (MBP) in vitro in the presence of [γ-32P]ATP (Fig. 1b). Taken together, these experiments indicate that the C-terminal part (residues 446-961) of LegK4 is dispensable for kinase activity.

LegK41–445 crystal structure.
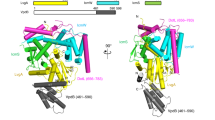
(a) Schematic representation of LegK41–445 protein sequence and the structurally assigned domains of LegK41–445. The “cap” domain (residues 1-65) is represented in pink, the kinase domain is separated in two lobes: the N-lobe (residues 66-148, green) and the C-lobe (residues 149-317, light blue) and the four-helix bundle (FHB) domain (residues 318-406) is coloured in yellow. (b) Purified LegK proteins were subjected to in vitro auto- and myelin basic protein (MBP) phosphorylation assays in the presence of [γ-32P]ATP. Phosphoproteins were separated by SDS-PAGE and visualised by autoradiography. (c) Ribbon representation of the crystal structure of LegK41–445 with domains coloured as in (a). (d) Topological diagram of LegK41–445, using the same colour code as in panels (a,c). The missing unstructured regions are represented as dashed lines.
Overall structure of LegK41–445
The structure of the apo form of LegK41–445 (apo-LegK41–445) was solved from a SeLegK41–445 crystal and refined at 3.7 Å resolution (Rwork/Rfree of 0.27/0.31) while the structure of substrate-bound LegK41–445 (named hereafter AMP-PNP•LegK41–445) was solved from a crystal grown in the presence of Mg2+ and AMP-PNP and refined at 2.5 Å resolution (Rwork/Rfree of 0.20/0.25) (Table 1 and Supplementary Fig. S1). Both apo and substrate-bound crystal forms contain two LegK41–445 molecules in the asymmetric unit (chains A1 and B1 for apo-LegK41–445 and A2 and B2 for AMP-PNP•LegK41–445). The overall root mean square deviation (rmsd) between A1 and B1 is 0.678 Å and 0.460 Å between A2 and B2.
The structure of AMP-PNP•LegK41–445 will be used to describe the general features of the structure of the protein. LegK41–445 is composed of three domains: a small N-terminal domain (residues 1 to 65), followed by the eukaryotic-like kinase domain itself consisting of two lobes, the N and C-lobe (residues 66 to 148 and 149 to 317, respectively) and a C-terminal domain (residues 321 to 406) (Fig. 1a,c,d). No density was observed for residues 407 to 445 suggesting that this part is disordered in the crystal.
The short amino terminal domain, named “cap” hereafter is well defined in chain A1, in which it consists of four small α-helices (α1 to α4) and a β-strand. βN1 runs antiparallel to the N-lobe strands β1 and β4 resulting in an extended six-stranded open β-barrel. The interactions between the cap domain and the N-lobe are extensive, burying a total of 970 Å2. A hydrophobic patch formed by α2 Leu22, Leu23 and Val26 and α3 Ile32 and Ile36 interacts with N-lobe residues Ile59, Phe61 (β1), Ile72 (β2), Tyr93 and Leu95 (β3). The interface also involves several hydrogen bonds including salt bridges between Asp56 and Arg46 and between Lys2 and Asp96 (Fig. 2a). Comparison of the cap domain in chains A1, A2, B1 and B2 reveals that parts of the cap domain are disordered as well as the contacting N-lobe elements (Fig. 2a,b). While no obvious homologue structure of this motif could be found using the DALI server, PDBefold26 identified several ubiquitin-associated domains (UBA) as remote structural homologues. UBA domains have been described in human AMPK-related kinases, among which are members of the MAP/microtubule affinity-regulating kinases family (MARK)27,28 (Fig. 2c). In this context, these short helical domains are C-terminal to the kinase domain and can regulate kinase activities through an interaction with the kinase N-lobe at β4 and the β2-β3 loop. Interestingly the cap domain is also similar to the N-terminal extension of the nuclear factor κB-inducing kinase (NIK). Like in LegK4, the extension of NIK also extends the N-lobe β-sheet. The extension of NIK also contains two extra helices, which stabilizes helix αC in the active orientation and maintains the kinase domain in the catalytically competent conformation29 (Fig. 2c). Although there are superficial similarities between the cap domain of LegK4 and UBA domains of MARK family kinases and the N-terminal extension of NIK, they bind to different faces of the N-lobe (Fig. 2a–c).

Structural flexibility of the cap domain.
Cartoon view of the interface between the cap domain (pink) and the N-lobe (green) of AMP-PNP•LegK41–445 (a) and apo-LegK41–445 (b). Side chains of principal residues participating in the interface in chain A2 are depicted as ball and sticks with carbon atoms coloured as the cartoon, oxygen in red and nitrogen in blue. (c) Structures of mouse NIK (pdb code 4G3C) and human MARK1 (pdb code 2HAK) kinases represented in the same orientation as LegK4 in (a) with the N-lobe coloured in green and the NIK N-terminal extension coloured in purple and MARK1 UBA domain coloured in magenta.
A structure-based sequence alignment of the LegK proteins suggests that the cap domain might be conserved in LegK1 and possibly in LegK2 but not in LegK3, which has a much shorter sequence (Fig. S1). The role of this domain is at present unclear in LegK4 but a LegK1 mutant deleted of the corresponding domain was inactive in vivo24. It is therefore likely that the cap domain fulfils a structural role in stabilizing the kinase N-lobe, similar to what has been previously observed with N-terminal extensions to kinase domains, such as the NIK29,30 or Nek731.
The kinase domain (KD) of LegK41–445 is very similar to other eukaryotic Ser/Thr kinases displaying the canonical N and C-lobes. The N-lobe contains a six-stranded antiparallel open β-barrel (βN1 and β1 to 5) and the αC α-helix. The C-lobe is composed of three antiparallel β-strands (β6-8) and six α-helices (αD to I). The KD of LegK41–445 contains the twelve conserved subdomains characteristic of eukaryotic kinases4. The KD is flanked by a four-helix bundle (FHB, α5 to α8) with the α5 inserted into a groove formed by αH and the loop αI-α5 (Fig. 1). FHB domains are ubiquitous and have numerous and divergent functions from secreted bacteriocins to human cytoskeleton binding proteins. Noteworthy a FHB domain is present in MAST1 (pdb code 2M9X), a member of the microtubule-associated Ser/Thr-protein kinase family but with no function yet associated. The sequence conservation for the FHB residues is weak amongst the LegK family but secondary structure prediction suggests that LegK1 and LegK2 might each have an all α-helical domain C-terminal to the KD (Supplementary Fig. S2). It is noteworthy that a mutant of LegK1 deleted of the C-term domain was still active for phosphorylation24. The position of the FHB in LegK4, i.e away from the kinase domain also suggests that it is likely not involved in the kinase activity, as is the case for LegK1.
LegK4 displays an atypical active kinase domain
The KD of LegK41–445 contains all the conserved elements of eukaryotic-like kinases, including an activation loop and a GHI domain contrasting with the minimal KD of the effector kinase so far characterized (Fig. 3a, Supplementary Fig. S2). Moreover, the P+1 loop activation segment residues Gly229 and Thr230 forms the GT motif, characteristic of Ser/Thr kinases32. Compared to the eukaryotic kinases, the structure displays however key differences in functional motifs. The position of the αG is distinctive in LegK41–445, as it is distant from the activation loop of the enzyme (Fig. 3a). In its usual position, αG plays an important role by binding to the P+1 loop and providing a scaffold/interface for protein substrate binding6. The active site of LegK41–445 displays a disordered P-loop (or glycine-rich loop, residues 79 to 87).

Structure of apo-LegK41–445.
(a) Cartoon representation of the crystal structures of apo-LegK41–445, mouse PKA mutant R194A (pdb code 4DFY) and NleH1 (pdb code 4LRJ). The helices αC, αF and αG are coloured in purple, orange and brown, respectively. The P- loop is coloured in blue and the activation segment coloured as follow: catalytic loop (green), Magnesium-binding loop (yellow), activation loop (magenta) and P+1 loop (cyan). (b) Close-up view of the catalytic site of apo-LegK41–445 (left) and PKA mutant (right) coloured as in (a). A side chain of important residues are shown as ball and sticks and coloured as the cartoon (carbon), red (oxygen) and blue (nitrogen). Main hydrogen bonds are indicated as dashed lines.
Remarkably, the activation segment of LegK41–445 is ordered (Fig. 3b). In most kinases, the activation segment is disordered in absence of phosphorylation as illustrated by the structure of a PKA mutant (R194A) that does not autophosphorylate on its activation loop33 (Fig. 3b). Ordering of the activation segment in many kinases requires phosphorylation of a serine or threonine residue in the activation loop. In the apo-LegK41–445 structure, a dense hydrogen bond network stabilizes the activation segment conformation (Fig. 3b). In particular, the catalytic loop residue Asp195 interacts with Asn200 and with Thr230 (P+1 loop) and His193 side chain binds to the hydroxyl group of Val212 (magnesium binding loop). The activation segment is further stabilized by Arg194 side chain which interacts with the main chain hydroxyl groups of Asp220 and Glu222, thereby connecting the catalytic and activation loops. In contrast to canonical protein kinases such as PKA, in which there is a short helix (αEF) between the APE motif at the C-terminal end of the activation loop and the start of αF, this region of LegK4 adopts an extended loop conformation that forms extensive stabilizing interactions with the activation loop. These include main chain interactions between Pro225 and Leu224 with Phe244 and Ile245. In addition, the position of the αC helix near the active site is stabilized via a hydrogen bond between its residue Glu115 and Trp228 (Fig. 3b).
These observations suggested that the protein might be in an active conformation without phosphorylation of the activation loop. To test this hypothesis, we searched for phosphorylation site(s) in LegK4 and LegK41–445 by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). Proteins were incubated with ATP followed by trypsin digestion. Mass spectrometry identified residue Thr823 to be phosphorylated in LegK4 and residues Ser422, Ser426 and Thr433 in LegK41–445 (Table 2). Ser422, Ser426 and Thr433 residues belong to the unstructured region, which could not be observed in the crystal structures. Since these are not phosphorylated in the full length LegK4, it is likely that phosphorylation of these residues is non-specific. Neither in LegK4 nor in LegK41–445 was the activation segment found to be phosphorylated. These results strongly suggest that, in agreement with our structural observation, the kinase activity of LegK4 does not require phosphorylation of the activation segment.
Nucleotide binding to the active site
In the AMP-PNP•LegK41–445 structure, one AMP-PNP molecule and two Mg2+ ions were present per LegK41–445 monomer (Fig. 4a). The activation segment, including the catalytic loop, the magnesium binding loop, the activation loop and the P+1 loop, adopts almost the same conformation as in the apo form (overall rmsd = 0.4 Å) (Fig. 4a). The nucleotide binds to LegK41–445 in a canonical manner, between the N- and C-lobe, with the adenine moiety buried in a hydrophobic pocket formed by Phe102, Phe147, Ile148 and Val212. The N1 and N6 amino groups of the adenine are hydrogen bonded to the Ile148 amide and to the Phe147 carbonyl, respectively while the N3 and N7 amino groups interact with surrounding bridging water molecules. Interestingly, the adenine is stacked by the Phe102 side chain. In the vast majority of kinases, this position is occupied by an alanine as part of the consensus “VAIK” sequence. In the case of kinase suppressor of Ras (KSR) 1, mutation of this residue to phenylalanine ablates the binding of ATP34 and the pseudokinase RYK, which does not bind nucleotide, also has a phenylalanine at this position35. In contrast, the presence of a phenylalanine in this position in LegK4 seems to stabilize the adenine ring rather than preventing nucleotide binding. The two hydroxyls of the ribose moiety are hydrogen bonded to the Asp152 side chain and the Gly199 carbonyl. The α- and β-phosphates interact with the invariant Lys104 and the Gln89, respectively (Fig. 4a). The γ-phosphate does not make direct interaction with the protein. The invariant Asp213, from the magnesium-binding loop, chelates the primary activating Mg2+ ion that directly binds the β-phosphate and bridges the γ-phosphate via a water molecule. The secondary Mg2+ ion interacts with the Asn200 side chain and bridges the α- and β-phosphates (Fig. 4a).

Nucleotide binding to LegK41–445.
(a) Cartoon representation of superimposed active sites of apo-LegK41–445 (gold) and AMP-PNP•LegK41–445 (blue). Side chains of important residues are shown as ball and stick and coloured as the cartoon (carbon), red (oxygen) and blue (nitrogen). (b) Structural superimposition of apo-LegK41–445.(gold) and AMP-PNP•LegK41–445 illustrating the movement of the cap domain and the N-lobe. (c) Comparison of the architectures of the C- (yellow surface) and R-spines (red surface) in the apo (upper panels) and nucleotide bound states (lower panels) of LegK41–445 and mouse PKA mutant R194A (pdb code 4DFY).
Comparison of the apo- and AMP-PNP•LegK41–445 structures shows that upon ligand binding, the protein undergoes a significant conformational change. A ∼12° rotation of the N-lobe and the cap domain around the active site brings closer together the two lobes of the catalytic domain as in other kinases (Fig. 4a,b). The P-loop, which usually covers and stabilizes the phosphates of the bound nucleotide, is disordered in apo-LegK41–445 and is still not visible in the electron density maps of the nucleotide-bound structure (Fig. 4a,b). To our knowledge, the structure of LegK41–445 is the first reported structure in which the P-loop is flexible in both, the open and the closed forms of the kinase protein structure. Indeed, the P-loop does not appear to contribute to ATP binding and other interactions, such as with Gln89 and Lys104, are sufficient to stabilize the position of nucleotide phosphates. It has been suggested that P-loop conformational changes do not necessary reflect a flexible loop but rather rigid movements from one conformation to the other upon substrate binding36. The absence of an ordered P-loop suggests that this loop might play either a role in directing the specificity of LegK4 kinase activity, and/or a role in regulation of kinase activity. In either case the P-loop may be stabilized upon interaction with a target protein.
A key difference between the apo- and AMP-PNP•LegK41–445 structures is the αC helix. In the AMP-PNP•LegK41–445 structure, αC Glu119 interacts with the Lys104 from β4 strand, allowing the proper orientation of the AMP-PNP α- and β-phosphates (Fig. 4a). Accordingly, a mutant with the corresponding Lys in LegK1 (Lys121, strain Philadelphia-1) changed to alanine failed to induce NF-κB activation in vivo24. This interaction is accompanied by a change in αC secondary structure. Upon AMP-PNP binding αC is one turn longer at its N-terminal than in apo-LegK41–445. This rearrangement triggers the inward movement of the N-lobe towards the C-lobe by shortening the β3- αC loop (Fig. 4a,b).
Activation of kinases can also be assessed by the alignment of two sets of spatially conserved hydrophobic residues, termed the catalytic (C-) and regulatory (R-) spines37,38. In inactive kinases the C and R-spines are disassembled while in active, nucleotide bound kinases, the spines form parallel columns of hydrophobic residues sitting on the αF helix (Fig. 4c). These spines span organize the catalytic infrastructure by restricting inter-lobe movements. As seen in Fig. 4c, the C and R-spines of LegK41–445 are already parallel in the apo form suggesting that the kinase is active. Binding of AMP-PNP completes the C-spine but with minimal structural rearrangement of the column. These observations are in agreement with the constitutive activity of the kinase.
A novel dimeric form stabilizes essential catalytic elements
Both the apo and the substrate bound LegK41–445 crystal forms contain two molecules in the asymmetric unit forming a similar dimer. The interface is symmetric and buries around 900 Å2 as determined by the PDBePISA server39 (Fig. 5a). To determine if the dimer of LegK41–445 was relevant in solution we used inline size exclusion small angle X-ray scattering (SEC-SAXS)40. SAXS data indicates that the Rg of LegK41–445 is 36.1 Å with a Dmax of 111 Å (Fig. 5b, Supplementary Fig. S3). These values are in excellent agreement with the theoretical Rg and Dmax of respectively 34 Å and 111 Å, derived from the dimer observed in crystal structure. The small difference might be due to the disordered region of 45 residues present at the C-terminal portion of each monomer. Comparison of LegK41–445 experimental SAXS data with the theoretical curves obtained with the crystal structures demonstrate that the best fit was obtained with the dimer of apo-LegK41–445 (χ2 = 4.6) compared to the dimer of AMP-PNP•LegK41–445 (χ2 = 10.5) or to the apo-LegK41–445 monomer (χ2 = 41.1) (Fig. 5b). These data clearly show that in solution, LegK41–445 adopts the conformation observed in the crystal structure.

LegK41–445 dimer organisation.
(a) Side (upper panel) and top (lower panel) views of the dimer with chain A coloured in grey and shown as a surface and chain B coloured in blue and shown as ribbon. The cap and FHB domains of the two chains are coloured in pink and yellow, respectively. (b) Comparison of the experimental SAXS curve of apo-LegK41–445 (black) with theoretical SAXS curves of apo-LegK41–445 monomer (orange), dimer (magenta) and AMP-PNP•LegK41–445 dimer (blue). χ2 obtained with FOXS server51 are indicated. (c) Detailed view of the dimer interface coloured as in (a) with participating residues shown as ball and stick.
The dimerisation involves predominantly charged residues from helices αF, αG and the αG-αI loop of the C-lobe and is essentially the same in the apo- and AMP-PNP•LegK41–445 structures (Fig. 5a,c). Briefly, αF Asp261 interacts with the backbone nitrogen of Val270 and Lys271 and Asn265 self interact. Reciprocal salt bridges occur between Glu262 and Lys271 residues. Interestingly, Ser232 located within the P + 1 loop forms a hydrogen bond with Asn163 from the αD-αE loop and this interaction occurs in both apo and substrate-bound forms (Fig. 5c). This stabilization role is usually played by helix αG, which is displaced here. The organisation of the dimer thus suggests that the assembly participates in the ordering of the activation loop in absence of phosphorylation. Dimerisation of kinases can promote activation through trans-phosphorylation or allosteric mechanisms (reviewed in6,41). Comparison of LegK41–445 dimer with the structures of other kinases that dimerise with the active sites facing each other, such as Chk242 and Ire143, reveals that the dimerisation mode of LegK4 is novel (Fig. 6a). Interestingly, upon binding to AMP-PNP, the dimer interface seems to act as a pivot onto which the two subunits close (Fig. 6d). Given the central part of the dimer obstructs part of the kinase surface that is usually involved in binding protein substrates, this feature might restrict the substrate size. Together with the observation that the P-loop remains disordered in the presence of nucleotide, which is instead stabilized by atypical residues such as Phe102 and Gln89, it is possible that LegK4 binds protein substrates in a different orientation than most kinases.

Structural comparison of kinase dimers.
(a) Cartoon view of the crystal structures of dimeric kinases Chk2 (pdb code 2HK9) and Ire1 (pdb code 3P23). Kinase domains are coloured in wheat and grey and specific domains are coloured in yellow (Chk2) and green (Ire1). AMP-PNP is represented as dark blue spheres in Ire1. (b) Cartoon representation of apo- and AMP-PNP•LegK41–445 illustrating the closing of the dimer assembly upon binding AMP-PNP (represented as dark blue spheres). Kinase domains are coloured in wheat and grey, cap domains in pink and FHB domains in yellow.
Concluding Remarks
Eukaryotic-like kinase effectors represent a common strategy for bacterial pathogens to subvert host cell defences. While bacterial kinase effectors identified so far contained minimal kinase domains10, the structure of LegK4 reported here presents the characteristics of eukaryotic regulatory kinases albeit with remarkable, original modifications. Notably, nucleotide binding does not involve the P-loop, which has a highly divergent sequence, but is instead mediated by unusual residues in nearby structural elements. The newly described dimer and atypical activation loop suggest a structural explanation for LegK4 constitutive activity that was unpredictable from sequence analysis. Our analysis indicates that some of these elements might be shared by LegK1 and possibly other members of the family. The structure also points toward specific mechanisms of kinase regulation within the family that could be exploited by the bacteria to re-route different host pathways. While a number of these questions remain open, our study has paved the way for detailed and mechanistic studies aiming at understanding the role of eukaryotic-like kinases during bacterial infection.
Experimental Procedures
Cloning, over-expression and protein purification
PCR was used to amplify the DNA fragment encoding for LegK4 (forward primer 5′-CACCATGAAATTGCCTTCGGTTTCATGAAT-3′, reverse 5′-TTAATATGGCAAAATGATGACGTTGC-3′) and LegK4 amino-acids 1 to 445 (reverse primer 5′-TTAATCTGAAACTTGTTTAATCGCTGC) and inserted into the pET151/D TOPO (Invitrogen) following manufacturer procedure. The DNA fragment coding for LegK4 amino-acids 1 to 445 was inserted into the pET151/D- to express the protein LegK41–445 in fusion with a N-terminal His6-tag followed by a tobacco etch virus protease (TEV) cleavage site. The resulting vectors were introduced into the E. coli BL21 (DE3) and the bacteria were cultured at 37 °C in Lysogeny broth medium supplemented with 100 μg/ml ampicillin up to an optical density at 600 nm of 0.6–0.9. Protein expression was induced by adding 1 mM Isopropyl β-D-1-thiogalactopyranoside and the bacteria were grown at 20 °C for 16 hours. Cells were harvested by centrifugation and flash frozen in liquid nitrogen before being stored at −80 °C. Frozen cells were re-suspended in lysis buffer (50 mM Tris, pH 8.0, 0.5 M NaCl, 10% Glycerol (V/V) and 20 mM Imidazole) supplemented with 5 U/ml of benzonase (Novagen) and a tablet of a Ethylenediamine tetra-acetic acid (EDTA)-Free protease inhibitors cocktail (Roche). Re-suspended cells were disrupted by sonication and centrifuged for 20 min at 16000 g at 4 °C. The supernatant was then loaded onto a HisTrap 5 ml column (GE Healthcare), previously equilibrated with 15 ml of equilibration buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 5% glycerol and 20 mM Imidazole). The column was washed with 20 ml of the equilibration buffer and 20 ml of equilibration buffer containing 50 mM Imidazole. The proteins were eluted with a 50–500 mM Imidazole gradient). The His6-tag of LegK4 and LegK41–445 were then cleaved by mixing a His6 tagged TEV protease and the fusion protein at 1:40 (w/w) ratio in the presence of 0.5 mM EDTA and 0.5 mM Dithiothreitol (DTT). The mixture was dialysed against 20 mM Tris, pH 8.0, 150 mM NaCl, 5% Glycerol (V/V) at 4 °C. Cleaved LegK4 or LegK41–445 were then separated from the uncleaved fraction and TEV protease by reloading the dialysed protein onto a Nickel affinity column. Both LegK4 and LegK41–445 were then further purified on an anion exchange column (HiTrap Q; 5 ml; GE Healthcare) equilibrated with 20 mM Tris, pH 8.0 and 5% glycerol. The proteins were eluted with a NaCl gradient (0 to 1 M). A final size exclusion chromatography step (HiLoad 16/600 Superdex 200 pg column; GE Healthcare) was performed in 20 mM Tris, pH 8.0, 150 mM NaCl, 5% Glycerol (V/V). Selenomethionine labelled LegK41–445 (SeLegK41–445) was produced following the protocol described in (13) and purified as LegK41–445.
Crystallization, data collection and processing
LegK41–445 and SeLegK41–445 were concentrated to 8–10 mg/ml using an Amicon Ultra 10-kDa concentrator. The vapour diffusion sitting drop technique was used for crystallization with drops, consisting of 1 μl protein and 1 μl reservoir buffer equilibrated against 500 μl of reservoir solution in 24-well plates (Intelli-plates, ARI) at 292K. Native LegK41–445 was co-crystallized with 1 mM Adenylyl imidodiphosphate (AMP-PNP) and 2 mM MgCl2, which were added to the protein 10–20 minutes prior to setting-up crystallization drops. Plate shaped crystals were obtained for LegK41–445 in complex with AMP-PNP, with a reservoir solution consisting of 10% (w/v) polyethylene glycol (PEG) 20000, 20% (w/v) PEG 550 mono-methyl ether, 30 mM MgCl2, 30 mM CaCl2 and 100 mM Tris/Bicine pH 8.3. Bipyramidal crystals for SeMet derivative LegK41–445 grew with a reservoir solution 12% (w/v) PEG 4000, 20% glycerol, 0.1 M MOPS/HEPES pH 7.5, 5 mM Hexaammine cobalt (III) chloride. Crystals were then flash-cooled and stored in liquid nitrogen. Data for native LegK41–445 and Se-LegK41–445 were collected at 100K at ID23EH2 (European Synchrotron Radiation Facility) and Proxima 2a (SOLEIL) beamlines, respectively. Reflections were indexed, integrated and scaled with the XDS program suite44. The high-resolution cut-offs were estimated according to CC1/245. Data statistics and parameters are summarized in Table 1. Crystals of Se-LegK41–445 and LegK41–445 belonged to space group P321 and P1211, respectively and contained two molecules per asymmetric unit.
Structure solution and refinement
A high-redundancy data set was collected on a single Se-LegK41–445 crystal at the K absorption edge wavelength (0.979 Å). The initial heavy-atom sites were located using AutoSol46 from the Phenix program suite47. Despite the limited resolution of 3.7 Å, the initial phases had a very good figure of merit (FOM) of 0.580 and an excellent experimental electron density map enabled us to manually build an initial model. The resulting model was then used for molecular replacement calculations using the monoclinic data set at 2.49 Å resolution with Phaser47, in which two monomers were positioned with a translation function Z-score of 9.7. The resulting electron density map was then subjected to the AutoBuild program, part of the Phenix program suite47. An initial model consisting of 526 residues in 12 chains and 269 water molecules was obtained. Both native and derivative models were completed with sessions of manual model building using Coot combined with model refinement using Phenix47. Representative pictures of the electron density map are shown in Supplementary Fig. S1. Figures were generated with Pymol48. The coordinates and structure factors of the structures of apo-LegK41–445 and AMP-PNP•LegK41–445 have been deposited in the protein data bank with PDB codes 5CLR and 5CKW, respectively.
Small angle X-ray scattering
SAXS data were recorded on beamline Swing at SOLEIL Synchrotron (Gif sur Yvette, France) at a wavelength of 1.003 Å on a 17 cm × 17 cm low-noise Aviex charge-coupled device detector positioned at a distance of 1800 mm from the sample, with the direct beam off-centered. The useful Q-range was 0.004–0.61 Å−1, where Q = 4πsinθ/λ is the scattering vector and 2θ is the scattering angle. 50 μl of LegK41–445 (10 mg.ml−1) in buffer S (50 mM Tris pH 8.0, 200 mM NaCl, Glycerol 5% (v/v), 1 mM DTT) were injected into a size-exclusion column (Agilent BioSec-3) using an Agilent© HPLC system cooled at 15 °C and eluted directly into the SAXS flow through capillary cell40. SAXS data were collected online throughout the whole elution time, with a frame duration of 2 s and a dead time between frames of 1 s. A first data set of 90 frames, collected before the void volume, were averaged to account for buffer scattering. A second data set was collected for the sample, from which the 10 frames corresponding to the top of the elution peak were averaged and used for data processing after baseline subtraction. Data were processed using the local application FOXTROT49 and analysed using PRIMUS50. Theoretical curves from the models were generated by FoXS51.
In vitro phosphorylation assay
In vitro phosphorylation of about 2 μg of purified recombinant protein was performed for 30 min at 37 °C in 20 μl of a buffer containing 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM DTT, 10 μM ATP and 5 μCi of [γ-32P]ATP (Perkin-Elmer, Courtaboeuf, France). In some assays, MgCl2 was replaced with MnCl2. In eukaryotic substrate phosphorylation assays, 1 μg of MBP was added. In each case, the reaction was stopped by the addition of an equal volume of 2X Laemmli loading buffer.
Sample preparation and Mass Spectrometry analysis
The purified LegK4 and LegK41–445 proteins were subjected to in vitro phosphorylation with non-radioactive ATP, as described in the phosphorylation assays section, prior to SDS-PAGE separation and mass spectrometry analysis. The separated proteins were subjected to in-gel reduction, carbamidomethylation and tryptic digestion. The phosphopeptides were isolated using TiO2 spin tips (TitansphereTM Phos-TiO kit) according to the manufacturer protocol except for the elution steps. Retained phosphopeptides were eluted with 50 μL of 0.15 M ammoniumhydroxide solution then with another 50 μl of 60% CH3CN and 40% 0.3 M ammoniumhydroxide solution. Enriched phospho samples were analyzed by nano LC-MS/MS using an Ultimate nanoLC (ThermoFisherScientific) and a nanoESI-QToF-MS/MS mass spectrometry system (Q-Star XL, Applied Biosystems). Peptide separation was performed on a C18 PepMap micro-precolumn (Dionex) and a C18 PepMap nanocolumn (Dionex) where solvent was 0.1% HCOOH in H2O/CH3CN (95/5) and a gradient of solvent B (0.1% HCOOH in H2O/CH3CN (20/80)) was applied. MS data were acquired using Analyst QS 1.1 software (Applied Biosystems). Screening for phosphorylated peptides was achieved with ProteinPilot 4.0 software (Applied Biosystems) using the Paragon database search algorithm against a home-made database including the sequence of LegK4 and LegK41–445.
Additional Information
How to cite this article: Flayhan, A. et al. The structure of Legionella pneumophila LegK4 type four secretion system (T4SS) effector reveals a novel dimeric eukaryotic-like kinase. Sci. Rep. 5, 14602; doi: 10.1038/srep14602 (2015).
Change history
03 February 2016
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Manning, G., Whyte, D. B., Martinez, R., Hunter, T. & Sudarsanam, S. The protein kinase complement of the human genome. Science 298, 1912–1934, 10.1126/science.1075762 (2002).
Cousin, C. et al. Protein-serine/threonine/tyrosine kinases in bacterial signaling and regulation. FEMS Microbiol Let 346, 11–19, 10.1111/1574-6968.12189 (2013).
Shi, L., Potts, M. & Kennelly, P. J. The serine, threonine, and/or tyrosine-specific protein kinases and protein phosphatases of prokaryotic organisms: a family portrait. FEMS Microbiol. Rev. 22, 229–253 (1998).
Hanks, S. K. & Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 9, 576–596 (1995).
Taylor, S. S., Keshwani, M. M., Steichen, J. M. & Kornev, A. P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Phil. Trans. R. Soc. B 367, 2517–2528, 10.1098/rstb.2012.0054 (2012).
Endicott, J. A., Noble, M. E. & Johnson, L. N. The structural basis for control of eukaryotic protein kinases. Annu. Rev. Bio. 81, 587–613, 10.1146/annurev-biochem-052410-090317 (2012).
Kornev, A. P., Taylor, S. S. & Ten Eyck, L. F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 105, 14377–14382, 10.1073/pnas.0807988105 (2008).
Krachler, A. M., Woolery, A. R. & Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Biol. 195, 1083–1092, 10.1083/jcb.201107132 (2011).
Canova, M. J. & Molle, V. Bacterial serine/threonine protein kinases in host-pathogen interactions. J. Biol. Chem. 289, 9473–9479, 10.1074/jbc.R113.529917 (2014).
Grishin, A. M., Beyrakhova, K. A. & Cygler, M. Structural insight into effector proteins of Gram-negative bacterial pathogens that modulate the phosphoproteome of their host. Protein Sci. 24, 604–620, 10.1002/pro.2636 (2015).
Grishin, A. M. et al. NleH defines a new family of bacterial effector kinases. Structure 22, 250–259, 10.1016/j.str.2013.11.006 (2014).
Kim do, J. et al. Helicobacter pylori proinflammatory protein up-regulates NF-kappaB as a cell-translocating Ser/Thr kinase. Proc. Natl. Acad. Sci. USA 107, 21418–21423, 10.1073/pnas.1010153107 (2010).
Pruneda, J. N. et al. E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 33, 437–449, 10.1002/embj.201386386 (2014).
Grishin, A. M. et al. Structural basis for the inhibition of host protein ubiquitination by Shigella effector kinase OspG. Structure 22, 878–888, 10.1016/j.str.2014.04.010 (2014).
Hervet, E. et al. Protein kinase LegK2 is a type IV secretion system effector involved in endoplasmic reticulum recruitment and intracellular replication of Legionella pneumophila. Infect. Immun 79, 1936–1950, 10.1128/IAI.00805-10 (2011).
Horwitz, M. A. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158, 1319–1331 (1983).
Roy, C. R., Berger, K. H. & Isberg, R. R. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28, 663–674 (1998).
Berger, K. H. & Isberg, R. R. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19 (1993).
Finsel, I. & Hilbi, H. Formation of a pathogen vacuole according to Legionella pneumophila: how to kill one bird with many stones. Cell. Microbiol. 10.1111/cmi.12450 (2015).
Allombert, J., Fuche, F., Michard, C. & Doublet, P. Molecular mimicry and original biochemical strategies for the biogenesis of a Legionella pneumophila replicative niche in phagocytic cells. Microbes Infect. 15, 981–988, 10.1016/j.micinf.2013.09.007 (2013).
Isberg, R. R., O’Connor, T. J. & Heidtman, M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24, 10.1038/nrmicro1967 (2009).
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N. & Isberg, R. R. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. USA 103, 18745–18750, 10.1073/pnas.0609012103 (2006).
Shohdy, N., Efe, J. A., Emr, S. D. & Shuman, H. A. Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc. Natl. Acad. Sci. USA 102, 4866–4871, 10.1073/pnas.0501315102 (2005).
Ge, J. et al. A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc. Natl. Acad. Sci. USA 106, 13725–13730, 10.1073/pnas.0907200106 (2009).
Michard, C. et al. The Legionella Kinase LegK2 Targets the ARP2/3 Complex To Inhibit Actin Nucleation on Phagosomes and Allow Bacterial Evasion of the Late Endocytic Pathway. mBio 6, 10.1128/mBio.00354-15 (2015).
Krissinel, E. & Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 60, 2256–2268, 10.1107/S0907444904026460 (2004).
Marx, A. et al. Structural variations in the catalytic and ubiquitin-associated domains of microtubule-associated protein/microtubule affinity regulating kinase (MARK) 1 and MARK2. J. Biol. Chem. 281, 27586–27599, 10.1074/jbc.M604865200 (2006).
Panneerselvam, S., Marx, A., Mandelkow, E. M. & Mandelkow, E. Structure of the catalytic and ubiquitin-associated domains of the protein kinase MARK/Par-1. Structure 14, 173–183, 10.1016/j.str.2005.09.022 (2006).
Liu, J. et al. Structure of the nuclear factor kappaB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 287, 27326–27334, 10.1074/jbc.M112.366658 (2012).
de Leon-Boenig, G. et al. The crystal structure of the catalytic domain of the NF-kappaB inducing kinase reveals a narrow but flexible active site. Structure 20, 1704–1714, 10.1016/j.str.2012.07.013 (2012).
Richards, M. W. et al. An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9. Mol. Cell 36, 560–570, 10.1016/j.molcel.2009.09.038 (2009).
Taylor, S. S., Radzio-Andzelm, E. & Hunter, T. How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. FASEB J. 9, 1255–1266 (1995).
Steichen, J. M. et al. Structural basis for the regulation of protein kinase A by activation loop phosphorylation. J. Biol. Chem. 287, 14672–14680, 10.1074/jbc.M111.335091 (2012).
Hu, J. et al. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. USA 108, 6067–6072, 10.1073/pnas.1102554108 (2011).
Murphy, J. M. et al. A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem. J. 457, 323–334, 10.1042/BJ20131174 (2014).
Saraste, M., Sibbald, P. R. & Wittinghofer, A. The P-loop—a common motif in ATP- and GTP-binding proteins. Trends in biochemical sciences 15, 430–434 (1990).
Kornev, A. P. & Taylor, S. S. Defining the conserved internal architecture of a protein kinase. Biochim. Biophys. Acta 1804, 440–444, 10.1016/j.bbapap.2009.10.017 (2010).
Ten Eyck, L. F., Taylor, S. S. & Kornev, A. P. Conserved spatial patterns across the protein kinase family. Biochim. Biophys. Acta 1784, 238–243, 10.1016/j.bbapap.2007.11.002 (2008).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797, 10.1016/j.jmb.2007.05.022 (2007).
David, G. & Perez, J. Combined sampler robot and high-performance liquid chromatography: a fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Cryst. 42, 892–900, 10.1107/S0021889809029288 (2009).
Lavoie, H., Li, J. J., Thevakumaran, N., Therrien, M. & Sicheri, F. Dimerization-induced allostery in protein kinase regulation. Trends Biochem. Sci. 39, 475–486, 10.1016/j.tibs.2014.08.004 (2014).
Lountos, G. T. et al. Structural characterization of inhibitor complexes with checkpoint kinase 2 (Chk2), a drug target for cancer therapy. J. Struct. Biol. 176, 292–301, 10.1016/j.jsb.2011.09.008 (2011).
Ali, M. M. et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 30, 894–905, 10.1038/emboj.2011.18 (2011).
Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800, 10.1107/s0021889893005588 (1993).
Karplus, P. A. & Diederichs, K. Linking crystallographic model and data quality. Science 336, 1030–1033, 10.1126/science.1218231 (2012).
Zwart, P. H. et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435, 10.1007/978-1-60327-058-8_28 (2008).
Adams, P. D. et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 58, 1948–1954. Epub 2002 Oct 1921 (2002).
DeLano, W. L. http://pymol.sourceforge.net. (2002). Date of access: 02/03/2008.
Foxtrot. can be downloaded from http://www.synchrotron-soleil.fr/Recherche/LignesLumiere/SWING. (2009). Date of access: 01/04/2013.
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 36, 1277–1282, 10.1107/S0021889803012779 (2003).
Schneidman-Duhovny, D., Hammel, M., Tainer, J. A. & Sali, A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J 105, 962–974, 10.1016/j.bpj.2013.07.020 (2013).
Acknowledgements
The project was supported by a FINOVI foundation (www.finovi.org) project. We acknowledge the use of the UMS3444/US8 platform protein science facility and thank I. Zanella-Cléon and V. Gueguen-Chaignon for their assistance. We thank Proxima2 and Swing Beamline staff from SOLEIL and ID23EH2 beamline staff from ESRF. This work was performed within the framework of the LABEX ECOFECT (ANR-11-LABX-0048) of Université de Lyon, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR).
Author information
Authors and Affiliations
Contributions
A.F. performed the biochemistry, crystallised and solved the crystal structures. C.B. performed and analysed SAXS experiments. N.B. and P.D. performed phosphorylation assays. L.T. conceived the work. A.F., L.T. and R.B. analysed the structures. R.B., A.F. and L.T. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Flayhan, A., Bergé, C., Baïlo, N. et al. The structure of Legionella pneumophila LegK4 type four secretion system (T4SS) effector reveals a novel dimeric eukaryotic-like kinase. Sci Rep 5, 14602 (2015). https://doi.org/10.1038/srep14602
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14602
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
