
Abstract
Liver cirrhosis but also portal vein obstruction cause portal hypertension (PHT) and angiogenesis. This study investigated the differences of angiogenesis in cirrhotic and non-cirrhotic PHT with special emphasis on the canonical (Shh/Gli) and non-canonical (Shh/RhoA) hedgehog pathway. Cirrhotic (bile duct ligation/BDL; CCl4 intoxication) and non-cirrhotic (partial portal vein ligation/PPVL) rats received either atorvastatin (15 mg/kg; 7d) or control chow before sacrifice. Invasive hemodynamic measurement and Matrigel implantation assessed angiogenesis in vivo. Angiogenesis in vitro was analysed using migration and tube formation assay. In liver and vessel samples from animals and humans, transcript expression was analyzed using RT-PCR and protein expression using Western blot. Atorvastatin decreased portal pressure, shunt flow and angiogenesis in cirrhosis, whereas atorvastatin increased these parameters in PPVL rats. Non-canonical Hh was upregulated in experimental and human liver cirrhosis and was blunted by atorvastatin. Moreover, atorvastatin blocked the non-canonical Hh-pathway RhoA dependently in activated hepatic steallate cells (HSCs). Interestingly, hepatic and extrahepatic Hh-pathway was enhanced in PPVL rats, which resulted in increased angiogenesis. In summary, statins caused contrary effects in cirrhotic and non-cirrhotic portal hypertension. Atorvastatin inhibited the non-canonical Hh-pathway and angiogenesis in cirrhosis. In portal vein obstruction, statins enhanced the canonical Hh-pathway and aggravated PHT and angiogenesis.
Similar content being viewed by others


Introduction
Chronic liver injury activates hepatic stellate cells (HSCs), which produce collagen and show increased contraction1. Fibrosis and increased contraction of contractile cells lead to portal hypertension and complications2. Additionally, neo-angiogenesis has been identified as a key mechanism in the progression of liver cirrhosis with portal hypertension3. Neo-angiogenesis occurs within the diseased liver and in the splanchnic vascular bed. The extrahepatic angiogenesis further worsens the portal pressure due to higher portal venous inflow and by opening venous collaterals3,4,5. Interestingly, extrahepatic angiogenesis also occurs in absence of cirrhosis, for example due to portal vein obstruction or thrombosis6,7,8. However, there is scant information about the differences between cirrhotic and non-cirrhotic portal hypertensive angiogenesis.
Statins decrease fibrosis and lower portal hypertension in animals and humans mainly by blunting the RhoA/Rho-kinase-pathway in myofibroblastic HSCs9,10,11,12,13,14,15,16. Interestingly, RhoA/Rho-kinase-pathway seems to also play a role in the non-canonical Hedgehog-signaling (Hh)17,18. This crosstalk between RhoA/Rho-kinase and Hh-pathway might be mediated by Shh and is Gli-independent (Fig. 1A).

Simplified canonical and non-canonical hegdehog pathway and their potential implication in portal hypertension.
(A) Statin inhibit RhoA activation by hindering its isoprenylation by depletion of geranylgeranyl-pyrophosphate (GGPP). RhoA seems also to play a role in the non-canonical Hedgehog-signaling (Hh). This crosstalk between RhoA/Rho-kinase and Hh-pathway might be mediated by Shh and is Gli-independent. The canonical Hh-pathway is activated by Hh ligands, which after several steps activates Gli. Gli enhances transcription of downstream target genes. (B) Statins decrease fibrosis and lower portal hypertension in animals and humans with liver cirrhosis by blunting the RhoA/Rho-kinase-pathway and downregulation of canonical and non-canonical Hh-pathway in myofibroblastic HSCs. The non-canonical Shh/RhoA-signaling seems to play a major role in the extrahepatic angiogenesis in cirrhosis, whereas Gli-2 is a predominant mediator of canonical Hh-pathway in non-cirrhotic portal hypertension potentially mediating angiogenesis and aggravation of portal hypertension.
In liver cirrhosis, also canonical Hh-pathway is activated19. In this pathway, the cell surface receptor Patched-1 inhibits Smoothened. When Hh ligands bind Patched-1, Smoothened translocates and activates Gli transcription factors. Previous studies described that Hh pathway leads to the progression of liver diseases. Thereby, Gli enhances transcription of downstream target genes, which activate HSCs and promote the survival of these fibrogenic and contractile cells19,20,21,22,23,24,25.
Interestingly, it has been described that statin treatment might decrease Hh activation26,27. However, it is unknown whether statins interfere with canonical or with the non-canonical Hh signaling and which is their role in angiogenesis induced by portal hypertension.
Therefore, we investigated (i) the pathophysiological differences in angiogenesis induced by cirrhotic or non-cirrhotic portal hypertension, (ii) the effects of statins on angiogenesis in cirrhotic and non-cirrhotic portal hypertension and (iii) their role on the canonical and non-canonical Hh-pathway in portal hypertension.
Results
Hemodynamic changes in rats with portal hypertension after statin treatment
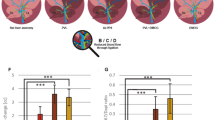
We compared two animal models of cirrhotic portal hypertension (BDL; CCl4-intoxication) with non-cirrhotic portal hypertension (PPVL). One week before sacrifice all animals received either atorvastatin-chow or control-chow (Fig. 2A). Atorvastatin reduced portal pressure in cirrhotic, CCl4-intoxicated rats, due to a significantly decreased hepatic vascular resistance compared to untreated animals (Fig. 2B,C). Splanchnic and systemic vascular resistance remained unchanged after atorvastatin treatment (Fig. 2D–F). Moreover, shunting was significantly decreased and accompanied by a slight increase in cardiac output (p = 0.0625) (Fig. 2E–G). By contrast, in non-cirrhotic PPVL rats, portal pressure increased compared to untreated PPVL rats, despite lower hepatic vascular resistance (Fig. 2B,C). This is accompanied by a significant decrease in splanchnic and systemic vascular resistance (Fig. 2D–F). Atorvastatin treatment led to a dramatic augmentation of shunt flow and cardiac output in PPVL rats compared to untreated PPVL rats (Fig. 2E–G).

In vivo hemodynamic measurements of CCl4-intoxicated and partial portal vein ligated (PPVL) rats with and without atorvastatin treatment.
(A) One week after PPVL, five weeks after BDL or fifteen weeks after CCl4-intoxication, rats received atorvastatin or control chow for 7 days before sacrifice. (B) Portal pressure was decreased in CCl4 rats and significantly upregulated in PPVL rats. (C) Hepatic vascular resistance was significantly downregulated in CCl4 as well as in PPVL rats. (D) Splanchnic vascular resistance remained unchanged in CCl4 rats and was significantly decreased in PPVL rats. (E) Shunt flow was decreased in CCl4 rats and significantly enhanced in PPVL rats after statin treatment compared to control. (F) Systemic vascular resistance was significantly reduced in PPVL rats and remained unchanged in CCl4-intoxicated rats. (G) In PPVL, as well as in CCl4-intoxicated rats, cardiac output was increased.
In summary, portal pressure, hepatic vascular resistance and shunt flow in cirrhosis were decreased by atorvastatin, as previously shown for the BDL model13. By contrast, they were enhanced in non-cirrhotic portal hypertension.
Hh signaling and profibrotic markers in human and rat portal hypertension
Since atorvastatin decreased hepatic vascular resistance in portal hypertension, we compared the expression of Hh-signaling in atorvastatin-treated animals with untreated animals.
It has been shown, that Shh and Gli are increased in cirrhosis, as well as RhoA/Rho-kinase-pathway. Interestingly, in human cirrhotic liver samples Sonic hedgehog (Shh) and Glioma associated oncogen family zink finger-2 (Gli-2) mRNA levels were increased (Fig. 3A). Similarly, Shh and Gli-2 protein expression was significantly upregulated in cirrhotic livers (Fig. 3B,C). Furthermore, the Hh components Shh and Gli-2 were significantly downregulated in cirrhotic livers of BDL and CCl4 – intoxicated rats after atorvastatin treatment (Fig. 3D). The mRNA levels of the profibrotic markers α-SMA, collagen-1, as well as vimentin were decreased after atorvastatin treatment in BDL and CCl4-intoxicated rat liver samples (Fig. 3E).

Hedgehog-signaling and profibrotic markers in human and rat livers.
(A) Shh and Gli-2 mRNA levels were upregulated in human cirrhotic liver samples and (B,C) protein expression levels of Shh and Gli-2 were significantly increased confirming previous data in animal models19,20,21,22,23,24,25, which were not repeated in this study. (D) Shh and Gli-2 mRNA levels were significantly downregulated in BDL and CCl4-intoxicated cirrhotic rat livers and significantly upregulated in PPVL liver samples after atorvastatin treatment. (E) The profibrotic markers α-SMA, Col1a1 and Vimentin were downregulated in BDL and CCl4-intoxicated rat livers after statin treatment and not affected in PPVL rat livers. In Fig. 3C the blots shown are from cropped nitrocellulose membranes, however the gel was not cropped and therefore have been run under the same experimental conditions. The cropping lines are shown in black.
By contrast, Shh mRNA levels were significantly increased in PPVL rats (Fig. 3D). Gli-2 mRNA levels were significantly upregulated in atorvastatin treated PPVL rats compared to the control group (Fig. 3D). mRNA levels of Hedgehog interacting protein (Hhip), a Hh-inhibitor, were significantly downregulated in PPVL rats and secreted frizzled-related protein 1 (sFRP1), a downstream target of Gli-2, was highly upregulated after atorvastatin treatment (Fig. 3D). In PPVL rats, the α-SMA, collagen-1 and vimentin levels were not altered by atorvastatin (Fig. 3E). Hh-signaling is enhanced in liver cirrhosis and atorvastatin blunts Hh-pathway together with profibrotic factors. By contrast, in the livers of PPVL rats, atorvastatin enhanced the Hh-pathway, which might be associated with increased levels of Hh ligands into circulation.
In vitro analyses of primary HSCs and human derived LX-2 cells
These cells are crucially involved in fibrosis and PHT. Cultured primary rat HSCs, were incubated with different doses of atorvastatin (10−4 M, 10−5 M and 10−6 M). The rational for using scratch assay to investigate the migration is that the cell-ECM and cell-cell interactions are still intact compared to other methods such as Boyden Chamber28. Interestingly, HSC migration as measured by scratch assay was significantly blunted by atorvastatin incubation (Fig. 4A,B). mRNA levels of the profibrotic markers α-SMA and collagen-1 were significantly downregulated after atorvastatin treatment in primary HSCs (Fig. 4C) and atorvastatin induced a significant decrease of Shh and sFRP1 (Fig. 4D).

In vitro analysis of primary rat HSCs and human derived LX-2 cells.
(A,B) HSCs had a reduced migration capability after incubation with atorvastatin. (C) α-SMA and Col1a1 mRNA levels were significantly reduced in HSCs incubated with atorvastatin. (D) Shh, Gli-2 and sFRP1 mRNA expression was downregulated in atorvastatin incubated HSCs. (E) Constitutive acitve (CA) RhoA increased Shh protein expression levels and cyclopamine inhibited this effect. (F) α-SMA mRNA levels in constitutive active RhoA transfected LX-2cells were upregulated and cyclopamine reversed this effect.
To differenciate between canonical and non-canonical Hh-signaling, we investigated statin effects on RhoA/Rho-kinase-pathway in activated HSCs. Human derived LX-2 cells transfected with a constitutively active (CA) RhoA plasmid had significantly increased mRNA and protein expression levels of Shh, compared to control cells and cells transfected with dominant negative (Dn) RhoA (Fig. 4E,F), while Gli-2 expression remained unchanged (data not shown). Incubation with cyclopamine, an inhibitor of the canonical Hh-pathway, reversed these effects (Fig. 4E).
The angiogenic and profibrotic potential of activated HSCs, as well as Hh-signaling is blunted by statins. This effect is mainly due to the non-canonical pathway, where Gli-2 expression has not been significantly decreased and Shh protein expression was upregulated by constitutive active RhoA.
Angiogenesis assays in vivo
Since shunt flow was reduced in cirrhotic and enhanced in non-cirrhotic portal hypertensive animals as compared to controls, we investigated the role of angiogenesis in extrahepatic vessels. Matrigels (collagen plug) were implanted for the investigation of angiogenesis in cirrhotic and non-cirrhotic portal hypertension. Intraperitoneal and subcutaneous matrigels were immunohistochemically stained for endothelial cells (ECs) with an antibody against CD31 and for vascular smooth muscle cells (VSMCs) with an α-SMA antibody. Similar to the hemodynamic changes, sprouting of new vessels in matrigel was decreased in cirrhotic portal hypertension (BDL/CCl4) by atorvastatin, whereas it was enhanced in non-cirrhotic portal hypertension (PPVL) by atorvastatin treatment (Fig. 5A,D; Suppl. Fig. 1A–D).

Angiogenesis assays in vivo.
(A) CD31 positive cells were reduced in intraperitoneal implanted matrigels of BDL and CCl4-intoxicated rats and enhanced in PPVL rats. (B) In BDL and CCl4-intoxicated rats, α-SMA positive cells were decreased in intraperitoneal implanted matrigels and upregulated in PPVL rats. (C) CD31+ cells were decreased in subcutaneous implanted matrigels of BDL and CCl4-intoxicated rats after atorvastatin treatment and upregulated in PPVL rats. (D) After atorvastatin treatment the amount of α-SMA positive cells in subcutanous implanted matrigel was decreased in Sham, BDL and CCl4-intoxicated rats and remained unchanged in PPVL rats.
Interestingly, atorvastatin also reduced VSMC migration in subcutaneously implanted matrigels (Fig. 5D; Suppl. Fig. 1D), which cannot be explained by shear stress due to portal hypertension in cirrhosis.
By contrast, atorvastatin treatment enhanced intraperitoneal migration of VSMCs (α-SMA) and ECs (CD31) into the matrigel in PPVL rats (Fig. 5A,B; Suppl. Fig. 1A,B). Furthermore, the subcutanous migration of ECs (CD31) inside the matrigel was enhanced in atorvastatin treated PPVL rats but remained unchanged for VSMCs (α-SMA) (Fig. 5C,D; Suppl. Fig. 1C,D). Atorvastatin decreased subcutaneous VSMC migration in Sham rats (Fig. 5D; Suppl. Fig. 1D).
Since, similarly to the activation of Hh-signaling in the respective livers, angiogenesis is blunted in cirrhosis after atorvastatin treatment, but enhanced in non-cirrhotic portal hypertension, we investigated the role of canonical and non-canonical Hh-signaling in the extrahepatic vessels.
Analysis of extrahepatic vessels
We evaluated the expression of Hh-signaling pathway in hepatic arteries of cirrhotic and healthy humans, as well as in the matrigel of the portal hypertensive animals after treatment with atorvastatin. Non-canonical Hh (Shh) was highly upregulated in cirrhotic hepatic arteries compared to non-cirrhotic arteries (Fig. 6A). In contrast, Gli2 mRNA levels were not affected (Fig. 6A), which might suggest the activation of non-canonical Hh-signaling. The explanted matrigels were analyzed using RT-PCR. Unchanged Gli-2 expression indicated that atorvastatin treatment did not alter canonical Hh-signaling, while downregulation of Shh abolished the non-canonical variant in subcutaneous and intraperitoneal implanted matrigels of BDL and CCl4-intoxicated rats (Fig. 6B,C). Interestingly, canonical Hh-pathway (Gli-2) was highly upregulated in PPVL rats treated with atorvastatin in both, intraperitoneal and subcutaneous implanted matrigels (Fig. 6B,C). The migration of VSMC in vitro was inhibited by atorvastatin incubation, similarly to the effect of atorvastatin on VSMC migration into subcutaneous matrigel of Sham rats (Fig. 6D). Moreover, the inhibition of canonical Hh-pathway using cyclopamine had no effect on the migration of VSMCs in vitro. By contrast, human umbilical vein endothelial cells (HUVECs) formed significantly more tubes after incubation with atorvastatin in vitro.

Analysis of extrahepatic vessels.
(A) Shh mRNA levels were highly upregulated in hepatic arteries of liver transplant recipients compared to donors. (B) Shh mRNA levels were downregulated in subcutaneous implanted matrigel of BDL rats after atorvastatin treatment. Gli-2 was highly upregulated in subcutaneous implanted matrigels of PPVL rats. (C) Shh mRNA levels were reduced in intraperitoneal implanted matrigel of BDL and CCl4-intoxicated rats after statin treatment and Gli-2 was highly upregulated in PPVL rats. (D) Atorvastatin reduced migration of A7r5 rat aortic VSMCs significantly in vitro. Incubation with cyclopamine had no effect on migration capability of VSMCs. (E,F) Endothelial cells (HUVEC) formed more tubes after incubation with atorvastatin in tube formation assay.
In summary, these data suggest a greater role of Shh/RhoA-signaling in the extrahepatic angiogenesis in cirrhosis, whereas Gli-2 is a predominant mediator of canonical Hh-pathway in non-cirrhotic portal hypertension (Fig. 1B).
Discussion
This study shows for the first time, that the mechanisms of neo-angiogenesis in cirrhotic and non-cirrhotic portal hypertension are different. Statin therapy blunts neo-angiogenesis in liver cirrhosis, probably due to an inhibition of non-canonical Hedgehog signaling among other mechanisms. Furthermore, in non-cirrhotic portal hypertension, statins increase splanchnic and systemic angiogenesis, together with an upregulation of the canonical Hedgehog pathway activity.
In previous studies we showed that atorvastatin significantly blunts portal hypertension and fibrogenesis in BDL rats13,14 and that statins inhibit proliferation and apoptosis and induce senescence in hepatic myofibroblasts15. Our present study confirms these results in a second model of fibrosis (CCl4-intoxication): also in the CCl4-induced cirrhosis, the profibrotic markers α-SMA and collagen-1 were decreased after statin treatment.
Hedgehog pathway activation has previously been identified as a cause of activation of hepatic stellate cells and in consequence contraction and production of extracellular matrix29. Indeed, the present study clearly shows an upregulation of the Hedgehog pathway, associated with progression of liver fibrosis. Moreover, statin treatment reduced Hedgehog pathway activity in cirrhotic livers and in activated hepatic stellate cells. This might be a consequence of the previously suggested non-canonical mechanisms of Hedgehog downstream activation, via RhoA/Rho-kinase17,30, since atorvastatin inhibits RhoA13,14, which is clearly interacting with Hh-signaling as shown by our gain of function experiments in HSCs. At least partly by this mechanism, statins decrease Hedgehog pathway activity in cirrhotic rat livers. Moreover, statin treatment may lead to inhibition of canonical Hedgehog pathway, shown by decreased Gli-2 expression. This probably is a further explanation for the previously described beneficial effects of statins on liver fibrosis13,14,15.
In cirrhotic BDL and CCl4-intoxicated rats not only fibrosis was blunted by statins, but also the intrahepatic vasoconstriction was significantly reduced13,31. In line with these previously published findings from others and our group, a significant decrease in portal pressure in BDL rats and a trend towards lower portal pressure in CCl4-intoxicated rats after treatment with atorvastatin was reported13,31. This difference between the models might be caused by a more aggressive, cholestatic injury induced by the BDL model.
Portal hypertension leads to increased neo-angiogenesis and thereby enhances portal-systemic shunting3. Our current study not only confirmed previous finding in BDL rats that the hepatic shunt flow was reduced in atorvastatin-treated rats13, but also extended this finding to the CCl4-model. Summarizing previous and current results, atorvastatin treatment decreased fibrogenesis, hepatic resistance and shunt flow in both cirrhotic models in rats.
Moreover, statins reduced neo-angiogenesis in the splanchnic circulation. The decreased splanchnic neo-vascularization in statin-treated cirrhotic animals may be due to the decreasing effects of atorvastatin on fibrosis and portal pressure. Thereby, shear stress in splanchnic vessels is reduced and one of the major driving forces for angiogenesis in portal hypertension is blunted. However, atorvastatin blunted the neo-vascularization of subcutaneously implanted matrigels in the cirrhotic rats. Therefore, these results cannot only be explained by the role of portal pressure, but also by the general anti-angiogenic effect of statins, that might be mediated by Hedgehog signaling. Indeed, Hedgehog pathway is crucially involved in angiogenesis, tumor vascularization and remodeling in diseased livers32,33,34. The reduced Hedgehog pathway activity in cirrhotic livers may affect the splanchnic angiogenesis via influencing endothelial and vascular smooth muscle cells. Interestingly and similar to the findings in rats, in human cirrhosis the Hedgehog ligands, but not their effector Gli-2, are upregulated. Therefore, a possible explanation could be the inhibition of circulating pro-angiogenetic factors, as well as Hedgehog components, which in cirrhosis generally show increased expression and are released into the circulation.
Statin treatment of non-cirrhotic portal hypertensive rats led to surprising and contrary results. In these rats atorvastatin increased portal pressure and shunt flow, probably due to highly upregulated neo-angiogenesis in peritoneal and subcutaneous vessels. Contrary to its effect in hepatic and cirrhotic vessels, statin treatment enhanced the activation of Hedgehog pathway in splanchnic and systemic vessels of PPVL rats. Fibrosis, inflammation and portal hypertension in cirrhosis might lead through different cytokines and chemokines to extrahepatic vascular changes. Since atorvastatin ameliorates fibrosis and portal hypertension, these liver derived effects are less pronounced after atorvastatin treatment. In PPVL the liver is not cirrhotic and the hepatic effects of statins mediated by modulation of cirrhosis are lacking. This might be a possible explanation, that atorvastatin has opposing effects in PPVL rats due to a non-canonical Hedgehog regulation. Furthermore, this study showed that different molecular mechanisms underlie angiogenesis in cirrhotic and non-cirrhotic portal hypertension.
In conclusion, the broad use of statins and their recently recognized beneficial effect on liver cirrhosis and portal hypertension9,35,36 point towards a potential clinical relevance of this study. While our data present further evidence for the beneficial use of statins in liver cirrhosis, caution is required in non-cirrhotic portal hypertension. Further studies are warranted, to investigate the different cellular mechanisms regulating angiogenesis in cirrhotic and non-cirrhotic portal hypertension.
Materials and Methods
Animals
55 wild type (WT) rats (Sprague Dawley) were used for our experiments. All experiments were performed in accordance with relevant guidelines and regulations approved by LANUV, the responsible committee for animal studies in North Rhine-Westphalia (permission number 8.87-50.10.31.08.287). Sprague Dawley rats were housed in a controlled environment (12 hour light/dark, temperature 22 °C to 24 °C) and fed standard rat chow (Ssniff, Soest, Germany).
Model of cirrhotic portal hypertension
Cholestatic model of cirrhosis
Bile duct ligation (BDL) was performed in nine WT rats with an initial body weight (BW) of 180–200 g as described previously13,37. Experiments were carried out six weeks after BDL. Eight sham-operated rats served as controls. Five rats undergoing BDL for six weeks received atorvastatin (15 mg/kg BW per day) on the last seven days before sacrifice, as described previously13.
Toxic model of cirrhosis
20 rats with an initial BW of 100–120 g twice weekly underwent inhalation of 1l/min CCl4 for 16 weeks until ascites, as a definite sign of portal hypertension, was present as described previously37. Seven rats undergoing CCl4-intoxication for 16 weeks received atorvastatin (15 mg/kg BW per day) on the last seven days before sacrifice.
Model of non-cirrhotic portal hypertension
Partial portal vein ligation (PPVL)
PPVL was performed in 18 WT rats with an initial BW of 180–200 g as described previously38,39. Experiments were carried out two weeks after PPVL. Seven rats undergoing PPVL for two weeks received atorvastatin (15 mg/kg BW per day) on the last seven days before sacrifice38,39.
In vivo hemodynamic experiments
In vivo studies were performed in eleven WT PPVL rats, 13 WT CCl4 and nine WT BDL rats and their respective controls. Rats were used for the hemodynamic studies as described previously40. To assess the effects of atorvastatin, invasive measurements of mean arterial pressure (MAP) and portal pressure (PP) were performed in cirrhotic rats.
Microsphere technique
To investigate hemodynamics, the colored microsphere technique was performed as described previously40. 300.000 systemic (red) microspheres (15 μm diameter, Triton-Technologies, San Diego, USA) were injected in the left ventricle. Mesenteric portal-systemic shunt volume was estimated by injection of 150.000 microspheres (yellow) in the ileocecal vein40.
Human liver samples
The Human Ethics Committee of the University of Bonn (202/01) approved the use of human liver and hepatic arteries, taken during liver transplantation from patients with alcohol-induced cirrhosis (n = 5). Liver and hepatic artery samples from non-cirrhotic patients submitted to liver resection served as controls (n = 5). The methods were carried out in accordance with the approved guidelines. An informed consent was obtained from all patients. No patient or donor received catecholamines, ACE inhibitors or angiotensin receptor antagonists before transplantation37. Samples were snap frozen following excision.
Western blotting
Liver samples were processed as previously described using SDS-PAGE gels and nitrocellulose membranes13,15. Ponceau-S staining assured equal protein loading. GAPDH served as endogenous controls. Membranes were incubated with the respective primary antibody (Supplementary Table 1) and corresponding secondary peroxidase-coupled antibody (Santa-Cruz-Biotechnology, Santa Cruz, USA). After enhanced chemiluminescence (ECL, Amersham, UK) digital detection was evaluated using Chemi-Smart (PeqLab Biotechnologies, Erlangen, Germany).
Quantitative RT-PCR
RNA isolation, reverse transcription and detection by RT-PCR were performed as described previously13,37. Assays provided by Applied Biosystems (Foster City, USA) are listed in Supplementary Table 2. 18S rRNA served as endogenous control. Results of HSCs and liver samples were expressed as 2−ΔΔCT and express the x-fold increase of gene expression compared to the control group.
Immunohistochemical staining for CD31 and α-SMA
Stainings for CD31 and α-SMA were performed in cryosections from liver tissue (3 and 7 μm thick, respectively) as described previously41,42,43. Briefly, after several steps cryosections were incubated with a mouse-anti-SMA antibody (clone 1A4; Sigma-Aldrich, Munich, Germany), or with antibody against CD31 (ab24590, Abcam, Cambridge, UK). Thereafter, a biotinylated rabbit-anti-mouse secondary antibody (Dako, Glostrup, Denmark) was used.
Isolation of primary HSCs
Rat HSCs were isolated as described previously37,44. Briefly, primary HSCs were isolated in a two-step pronase-collagenase perfusion from the livers of WT rats and fractionated by density gradient centrifugation. Viability and purity were systematically over 95%. Cells were seeded on uncoated plastic culture dishes. Experiments were performed seven days after isolation or after the first passage (10 days) when HSCs were fully activated.
Incubation with atorvastatin
Atorvastatin (10−6 M, 10−5 M, 10−4 M) was added to the culture medium of these cells as indicated for three days, or cells remained untreated.
Transfection with RhoA plasmids
LX-2 cells were kindly provided by our co-operation partner Vijay H. Shah (Mayo Clinic, Rochester, USA)45. Twenty-four hours before transfection, 6 × 105 LX2 cells were incubated with transfection media (Dulbecco’s modified Eagle’s medium [DMEM] with 10% fetal calf serum [FCS] without penicillin/streptomycin). The RhoA plasmid (pEGFP-C2) and the respective empty control plasmid were isolated according to manufacturer instructions (NucleoBond Xtra Maxi kit; Machery, Nagel, Germany). Plasmid (15 μg) and 37.5 μL of lipofectamine were incubated for 20 minutes with a total volume of 3.6 mL of media. After removal of media the plasmid/lipofectamine mix was added drop-wise to cells. After 3–4 hours, mixture removed and cells were again incubated with fresh media, containing 10% FCS and harvested after 3 days. Efficacy of transfection was tested by their GFP-expression and confirmed by RT-PCR and Western blotting.
Scratch assays
To observe the cell migration a scratch assay was performed as described previously28. Briefly, primary rat HSCs or A7r5 VSMCs were cultured in 24-well plates until the wells were completely covered with cells. A line was scratched through the cell layer with a pipette tip (1 mm). Directly after scratching and at defined time points pictures were taken (Nikon, Eclipse 50i) and the scratch width was measured using the software NIS Elements D 3.2 (Zeiss, Germany).
Tube formation
To evaluate in vitro effects of atorvastatin on endothelial cells (HUVEC) (PromoCell, Heidelberg, Germany), a tube formation assay was performed as described previously46,47. A 24-well plate was coated with 300 μl Matrigel (Gibco/BRL, Karlsruhe, Germany) containing 10 ng/ml VEGF. After 24 h, 2.5 × 104 endothelial cells in 75 μl medium (Endothelial Cell Growth Medium+Supplemental Mix, PromoCell, Heidelberg, Cat.No. C-22010) were seeded in each well. Intercellular connections and tube-like formations were counted using a Nikon, Eclipse 50i microscope at 100× magnification.
Statistical analysis
Results are expressed as mean ± SEM unless otherwise indicated. Data were analyzed using Student’s unpaired and paired t-test where appropriate. Statistical analyses and graphing were performed using GraphPad Prism 4.0 for Macintosh. P < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Uschner, F. E. et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci. Rep. 5, 14573; doi: 10.1038/srep14573 (2015).
References
Friedman, S. L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 134, 1655–1669 (2008).
Sanyal, A. J., Bosch, J., Blei, A. & Arroyo, V. Portal hypertension and its complications. Gastroenterology 134, 1715–1728 (2008).
Bosch, J. & García-Pagán, J. C. Complications of cirrhosis. I. Portal hypertension. J. Hepatol. 32, 141–156 (2000).
Van de Casteele, M., Sägesser, H., Zimmermann, H. & Reichen, J. Characterisation of portal hypertension models by microspheres in anaesthetised rats: a comparison of liver flow. Pharmacol. Ther. 90, 35–43 (2001).
Groszmann, R. J. & Abraldes, J. G. Portal hypertension: from bedside to bench. J. Clin. Gastroenterol. 39, S125–130 (2005).
Darwish Murad, S. et al. Etiology, management and outcome of the Budd-Chiari syndrome. Ann. Intern. Med. 151, 167–175 (2009).
Plessier, A. et al. Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology 51, 210–218 (2010).
Seijo, S. et al. Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology 57, 1962–1968 (2013).
Abraldes, J. G. et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 136, 1651–1658 (2009).
Laufs, U. & Liao, J. K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 273, 24266–24271 (1998).
Laufs, U., Marra, D., Node, K. & Liao, J. K. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors attenuate vascular smooth muscle proliferation by preventing rho GTPase-induced down-regulation of p27(Kip1). J. Biol. Chem. 274, 21926–21931 (1999).
Rikitake, Y. & Liao, J. K. Rho GTPases, statins and nitric oxide. Circ. Res. 97, 1232–1235 (2005).
Trebicka, J. et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 46, 242–253 (2007).
Trebicka, J. et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 53, 702–712 (2010).
Klein, S. et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab. Invest. 92, 1440–1450 (2012).
Marrone, G. et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut (2014). 10.1136/gutjnl-2014-308338.
Chinchilla, P., Xiao, L., Kazanietz, M. G. & Riobo, N. A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9, 570–579 (2010).
Renault, M.-A. et al. Sonic hedgehog induces angiogenesis via Rho kinase-dependent signaling in endothelial cells. J. Mol. Cell. Cardiol. 49, 490–498 (2010).
Omenetti, A., Choi, S., Michelotti, G. & Diehl, A. M. Hedgehog signaling in the liver. J. Hepatol. 54, 366–373 (2011).
Beachy, P. A., Hymowitz, S. G., Lazarus, R. A., Leahy, D. J. & Siebold, C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 24, 2001–2012 (2010).
Choi, S. S. et al. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 285, 36551–36560 (2010).
Jung, Y. et al. Signals from dying hepatocytes trigger growth of liver progenitors. Gut 59, 655–665 (2010).
Omenetti, A. & Diehl, A. M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 27, 268–275 (2011).
Omenetti, A. et al. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab. Invest. 87, 499–514 (2007).
Yang, L. et al. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 48, 98–106 (2008).
Beachy, P. A. et al. Multiple roles of cholesterol in hedgehog protein biogenesis and signaling. Cold Spring Harb. Symp. Quant. Biol. 62, 191–204 (1997).
Cooper, M. K., Porter, J. A., Young, K. E. & Beachy, P. A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603–1607 (1998).
Liang, C.-C., Park, A. Y. & Guan, J.-L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2, 329–333 (2007).
Choi, S. S. et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G1093–1106 (2009).
Maitah, M. Y., Ali, S., Ahmad, A., Gadgeel, S. & Sarkar, F. H. Up-regulation of sonic hedgehog contributes to TGF-β1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS ONE 6, e16068 (2011).
Abraldes, J. G. et al. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J. Hepatol. 46, 1040–1046 (2007).
Xie, G. et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 62, 299–309 (2013).
Pola, R. et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med. 7, 706–711 (2001).
Pinter, M. et al. Hedgehog inhibition reduces angiogenesis by downregulation of tumoral VEGF-A expression in hepatocellular carcinoma. United European Gastroenterol J 1, 265–275 (2013).
Zafra, C. et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 126, 749–755 (2004).
Simon, T. G., King, L. Y., Zheng, H. & Chung, R. T. Statin use is associated with a reduced risk of fibrosis progression in chronic hepatitis C. J. Hepatol. (2014). 10.1016/j.jhep.2014.08.013.
Trebicka, J. et al. Role of β3-adrenoceptors for intrahepatic resistance and portal hypertension in liver cirrhosis. Hepatology 50, 1924–1935 (2009).
Resch, M. et al. Alterations in mechanical properties of mesenteric resistance arteries in experimental portal hypertension. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G849–857 (2009).
Wiest, R. et al. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. J. Clin. Invest. 104, 1223–1233 (1999).
Grace, J. A. et al. Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology 145, 874–884 .e5 (2013).
Trebicka, J. et al. Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB2 receptor-deficient mice and decreased in CB1 receptor knockouts. Liver Int. 31, 860–870 (2011).
Huss, S. et al. Development and evaluation of an open source Delphi-based software for morphometric quantification of liver fibrosis. Fibrogenesis Tissue Repair 3, 10 (2010).
Trebicka, J. et al. Expression of vasoactive proteins in gastric antral mucosa reflects vascular dysfunction in patients with cirrhosis and portal hypertension. Liver Int. (2014). 10.1111/liv.12613.
Wojtalla, A. et al. The endocannabinoid N-arachidonoyl dopamine (NADA) selectively induces oxidative stress-mediated cell death in hepatic stellate cells but not in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G873–887 (2012).
Granzow, M. et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology 60, 334–348 (2014).
Raskopf, E. et al. Toxic damage increases angiogenesis and metastasis in fibrotic livers via PECAM-1. Biomed Res Int 2014, 712893 (2014).
Schmitz, V. et al. Plasminogen fragment K1-5 improves survival in a murine hepatocellular carcinoma model. Gut 56, 271–278 (2007).
Acknowledgements
The authors thank G. Hack and S. Bellinghausen for excellent technical assistance. The study was supported by grants from Bonfor Foundation (to JT), the Deutsche Forschungsgemeinschaft (SFB TRR57 to JT), as well as from grants of H. J. & W. Hector Stiftung (to JT).
Author information
Authors and Affiliations
Contributions
J.T. initiated and coordinated the work. F.E.U., G.R., S.S.C., M.G., S.K., E.R., S.G. and J.T. performed experiments. F.E.U., G.R., S.S.C., M.G. and J.T. performed statistics. P.F.M.v.d.V. and D.O.F. provided essential materials. F.E.U., G.R., S.S.C., M.G., T.S., P.F.M.v.d.V., A.M.D. and J.T. drafted the manuscript. All authors critically discussed and corrected the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Uschner, F., Ranabhat, G., Choi, S. et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci Rep 5, 14573 (2015). https://doi.org/10.1038/srep14573
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14573
This article is cited by
-
Rho-kinase inhibitor coupled to peptide-modified albumin carrier reduces portal pressure and increases renal perfusion in cirrhotic rats
Scientific Reports (2019)
-
Novel Targets and Drug Development in Portal Hypertension
Current Hepatology Reports (2019)
-
A Nitric Oxide-Donating Statin Decreases Portal Pressure with a Better Toxicity Profile than Conventional Statins in Cirrhotic Rats
Scientific Reports (2017)
-
Use of Statins in Patients with Chronic Liver Disease and Cirrhosis: Current Views and Prospects
Current Gastroenterology Reports (2017)
-
Statins: the Panacea of Cirrhosis?
Current Hepatology Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
