
Abstract
Brachypodium distachyon is a new model plant closely related to wheat and other cereals. In this study, we performed a comprehensive analysis of hormone-regulated genes in Brachypodium distachyon using RNA sequencing technology. Brachypodium distachyon seedlings were treated with eight phytohormones (auxin, cytokinine, brassinosteroid, gibberelline, abscisic acid, ethylene, jasmonate and salicylic acid) and two inhibitors, Brz220 (brassinosteroid biosynthesis inhibitor) and prohexadione (gibberelline biosynthesis inhibitor). The expressions of 1807 genes were regulated in a phytohormone-dependent manner. We compared the data with the phytohormone responses that have reported in rice. Transcriptional responses to hormones are conserved between Bracypodium and rice. Transcriptional regulation by brassinosteroid, gibberellin and ethylene was relatively weaker than those by other hormones. This is consistent with the data obtained from comprehensive analysis of hormone responses reported in Arabidopsis. Brachypodium and Arabidopsis also shared some common transcriptional responses to phytohormones. Alternatively, unique transcriptional responses to phytohormones were observed in Brachypodium. For example, the expressions of ACC synthase genes were up-regulated by auxin treatment in rice and Arabidopsis, but no orthologous ACC synthase gene was up-regulated in Brachypodium. Our results provide information useful to understand the diversity and similarity of hormone-regulated transcriptional responses between eudicots and monocots.
Similar content being viewed by others


Introduction
Human diets greatly depend on grasses, which have applications as energy crops and sustainable energy sources. Rice (Oryza sativa) and maize (Zea mays) are commonly used as model plants for monocots, as their genome sequence and extensive genetic resources are available1. Draper et al. proposed Brachypodium (Brachypodium distachyon) as a novel model plant in 2001 because it has advantages such as a small genome size, short life cycle, small plant size and the ability to self-pollinate2. Furthermore, Brachypodium is more closely related to wheat [Triticum aestivum] and rye [Secale cereale] than rice and maize are, which makes it convenient for analysis of different grass groups using model systems1. The whole genome sequence of Brachypodium inbred line Bd21 was released in 2010 as the first genome sequence available for the Pooideae subfamily3. Recently, a comprehensive collection of full-length cDNAs was reported by Mochida et al.4. Therefore, fundamental resources for Brachypodium molecular biology are being rapidly developed. Recently, Priest et al. reported an analysis of global gene expression in Brachypodium in response to abiotic stress5. Large-scale gene expression data are essential resources for model systems. For Arabidopsis, The Arabidopsis Information Resource (TAIR: http://www.arabidopsis.org) and Arabidopsis e-FP browser6 integrate the public transcriptome data with other information to provide information useful for analysis of gene functions. Many tools for biological data mining, such as co-expression analysis (e.g., ATTED-II7), transcriptome network analysis (AtCAST8,9) and prediction of transcriptome dynamics in natural conditions10, depend on the availability of large-scale transcriptome data.
Plant growth and development depend on phytohormone-mediated regulation of gene expression. Auxins, cytokinins (CKs), gibberellins (GAs) and brassinosteroids (BRs) generally promote plant growth and greatly influence plant stature and organ size. In contrast, ethylene, abscisic acid (ABA), salicylic acid (SA) and jasmonate (JA) regulate stress-related responses and/or growth retardation. The comprehensive response to a hormone was first reported in the eudicot model plant Arabidopsis thaliana by the AtGenExpress project (http://atpbsmd.yokohama-cu.ac.jp)11. Hundreds of genes have been identified as being regulated by phytohormones. These comprehensive transcriptome data have been utilized in hundreds of studies of large-scale or gene-specific regulation of transcripts. Therefore, analyses of phytohormone-regulated transcriptomes in Brachypodium are essential for transcriptomic and genetic studies of this plant. Brachypodium shares about 80% of its genes with rice as homologs12 and is more closely related to rice than to Arabidopsis and other model plants. Analyses of phytohormone-regulated transcriptomes were performed in rice for auxin, CK, SA, ABA, JA and ethylene13 and for auxin, CK, GA, BR, ABA and JA14. Comparisons of the responses in Brachypodium and these other model plants will reveal both responses common to all plants and those specific to Pooideae.
GA and BR treatment generates almost no change in gene expression in wild-type Arabidopsis seedlings11; indeed, the endogenous GA and BR levels are too high (i.e., saturated) for exogenous hormones to induce gene expression. Therefore, BR- and GA-regulated genes in Arabidopsis have been identified using the BR- and GA-deficient mutants det2 and ga-1, respectively and treatment with brassinolide (BL; BR), GA3, or GA411. In this study, we found that the transcriptional regulation of BR-regulated genes in BR-treated det2 plants was negatively correlated with BR-biosynthesis inhibitor treatment using the AtCAST database (http://atpbsmd.yokohama-cu.ac.jp/atcast/html/genelist/A0036_A0081.html)9. These findings suggest that the application of hormone biosynthesis inhibitors (Brz220 for BR and Phx for GA) to generate hormone-deficient conditions is a practical means of identifying BR- or GA-regulated genes in Brachypodium.
In this study, we performed a comprehensive RNA-seq analysis of the transcriptional responses in Brachypodium to eight phytohormones; auxin, BR, CK, GA, SA, ABA, JA and ethylene. The compounds assayed included indole-3-acetic acid (IAA; auxin), trans-zeatin (tZ; CK), BL, Brz220 (Brz; inhibitor of BR synthesis), GA4, prohexadione-calcium (Phx: inhibitor of GA synthesis), SA, ABA, methyl jasmonate (MJ; JA) and 1-amino-cyclopropane-1-carboxylic acid (ACC; ethylene precursor). In addition, we applied combination treatments of BL and Brz220 (a BR-biosynthesis inhibitor) and GA4 and Phx (GA-biosynthesis inhibitor) to Brachypodium. Transcriptional regulation of genes in Brachypodium was compared to that in Rice and Arabidopsis thaliana.
Results
Determination of hormone treatment conditions and identification of hormone-regulated genes
We selected phytohormone-responsive marker genes in Brachypodium using the following procedure. Twenty-eight phytohormone-responsive genes from rice13 and Arabidopsis thaliana11,15 were selected and their sequences were subjected to a BLAST search for the most similar sequences in Brachypodium. Top-hit Brachypodium genes were used to examine transcriptional responses in Brachypodium (Table 1). We checked the transcriptional responses with RT-PCR. In total, 19 of 28 genes showed reproducible gene expression responses to one of the phytohormones or the inhibitors in more than two independent experiments. These 19 genes were selected as phytohormone-responsive marker genes in Brachypodium (Table 1). Next, hormone treatment conditions in Brachypodium were determined using these marker genes. We used 1, 3 and 10 μM for IAA, tZ and GA; 10, 30 and 100 μM for Phx, ABA, MJ, ACC, Brz220 and SA; and 1, 10, 100 and 1000 μM for BL. The concentrations of phytohormones and inhibitors used in the comprehensive transcriptome analysis were determined accordingly. The optimum concentration of each phytohormone (Table 2) was determined as that at which the marker gene showed the greatest change in response to treatment compared to mock treatment. After the determination of treatment conditions, the hormone treatment and sampling were repeated twice independently and the hormonal responses were confirmed against the transcriptional responses of the marker genes. Total RNAs extracted in two independent experiments were mixed and used in RNA-seq analysis.
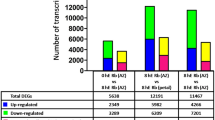
About 90% of obtained reads from the RNA-seq analysis were successfully mapped to the Brachypodium genome (Table 3). Differentially expressed genes (DEGs) in response to each treatment (FDR < 0.05) were analyzed using edgeR16 in a comparison with all other treatments (Table 4). Hundreds of genes were differentially expressed in response to ABA and MJ. Dozens of DEGs were detected for the IAA, tZ and SA treatments. We designated these analyses as being high stringency; the hormone-responsive genes are listed in Data S1–S8. In contrast, only four genes were differentially expressed in the Brz220 treatment. No DEG was detected in the mock 1-h, mock 3-h, Phx, Phx+GA, Brz220+BL and ethylene treatments, indicating that the gene expression responses to GA, BR and ethylene were weaker than those to the other hormones. Therefore, we conducted additional statistical analyses to identify the genes responsive to these hormones. For these analyses, DEGs were compared between the hormone treatment vs. two control treatments (Table 5). Two hundred and twenty-four DEGs were detected in response to GA, 474 DEGs in response to BR and 52 DEGs in response to ethylene. We designated these analyses as low stringency; the DEGs are listed in Table 4, Data S9–S16.
Comparison with rice and Arabidopsis
First, we compared the phytohormone-regulated genes in Brachypodium with those in rice. A comparison of our experimental conditions with those used in previous rice studies is presented in Table S1. Garg et al.13 reported the responses of rice seedlings to six hormones: auxin, CK, SA, ethylene, ABA and JA. We made a list of predicted orthologs of the Brachypodium DEGs in rice and evaluated whether they are regulated in the same manner (Table 6). Since Garg et al.13 reported phytohormone-regulated DEGs in rice without statistical information for each hormone treatment, we simply checked whether the orthologous genes from Brachypodium were included in the list of Garg et al.13 or not. We predicted 408 orthologs to the Brachypodium DEGs in rice. In total, 326 orthologous genes (80%) were regulated by phytohormones in the same manner. The transcriptional response to CK and ABA in Brachypodium was consistent with that in rice: 100% of the orthologs of the Brachypodium DEGs that were regulated by CK (tZ) were also regulated in rice by CK (BAP) and 98% of the orthologous genes were regulated by ABA in rice. Brachypodium and rice shared many auxin- and JA-responsive orthologous genes: 67% of the orthologs were also regulated by IAA in rice. Garg et al.13 reported that Aux/IAA and GH3 are IAA-responsive gene families. Consistent with this report, many genes in the Aux/IAA family (Bradi1g55370, Bradi2g04910, Bradi2g34030, Bradi2g16850, Bradi2g46420, Bradi2g31820, Bradi3g55410, Bradi4g35960, Bradi3g54610 and Bradi2g11120) and GH3 family (Bradi2g50840, Bradi1g22830 and Bradi2g52000) were found to be auxin-responsive in Brachypodium. In total, 42% of the orthologous genes were JA-responsive in rice. JAZ1-like genes in Brachypodium (Bradi5g24410), OsJAZ1, a MYC2-like gene in Brachypodium (Bradi3g34200) and OsMYC2 were regulated in both plants. Transcriptional responses to SA and ethylene were less conserved. Only 26% and 31% of the orthologous genes were regulated by SA and ethylene in rice. A transcription factor gene, OsWRKY45, which acts in SA-mediated pathogen resistance17 and its orthologous gene in Brachypodium (Bradi2g44270), were found to be regulated by SA in both plants. Ethylene receptor-like genes (Bradi5g00700 and LOC_Os04g08740) were found to be regulated in both plants. Sato et al.14 performed a transcriptional analysis using auxin, CK, GA, BR, ABA and JA and presented a coexpression database. Since they did not show phytohormone-regulated genes, we did not directly compare our hormone-induced gene expression results with those of Sato et al.14.
A gene ontology (GO) term enrichment (GOE) analysis was performed to evaluate the DEGs for Brachypodium. In this study, the GO terms for Brachypodium were redefined using the latest GO annotation for Arabidopsis thaliana (see the Methods for details) because the GO terms inferred from InterProScan (http://geneontology.org/page/go-annotation-standard-operating-procedures) lack useful GO terms for the study of phytohormone responses (e.g., the “Response to cytokinin stimulus”; Table S2). Therefore in the following sections, a detailed analysis was performed with the help of GO terms predicted from Arabidopsis (Tables 7 and 8). In this study, we analyzed the GOE for Arabidopsis using the latest GO terms (R package ath1121501cdf version 2.14.0) and microarray data provided by Goda et al.11 (Table S3) to compare the results of GOE analyses with those in Brachypodium.
Auxin response
One hundred and twenty-five genes were identified as auxin-responsive genes in the high-stringency analysis (Data S1). The genes with the term “response to auxin stimulus” were found to be enriched in the GOE analysis of auxin-responsive genes in Brachypodium (Table 7). As we described in the comparison with rice, many genes in the Aux/IAA family and GH3 family were detected as auxin-responsive genes in Brachypodium. These results are also consistent with the auxin response in Arabidopsis (Fig. 1, Fig. S1). In contrast to the auxin response in Arabidopsis, Brachypodium genes related to ethylene functions were not regulated in the same manner (Fig. S2), although their expression levels were not low (present at sufficiently high levels for analysis) (log2 cpm (number of counts per million reads) >1).

Phylogenic tree of Aux/IAA genes and their transcriptional responses to auxin in Brachypodium and Arabidopsis.
Brachypodium Aux/IAA family members were retrieved using the BLAST software. Protein sequences were aligned with MAFFT and similarities (percentage identity) are shown as a phylogenetic tree. Brachypodium genes are shown in red and Arabidopsis genes in black. log2 ratio represents the gene expression ratio between the read count in IAA treatment divided by the average read count from all other treatments. Stat represents the p-value of the microarray experiment of Arabidopsis treated with IAA11 or the FDR of RNA-seq data when the count of IAA treatment was compared with all other treatments. log2 ratio is hatched in red if genes are up-regulated. cpm represents the average of log2-scaled read counts per million reads in all experiments.
CK response
Twenty-three genes were identified as cytokinin-responsive genes in the high-stringency analysis (Data S2). The genes with the term “response to cytokinin stimulus” were enriched in the GOE analysis (Table 7). Genes in the type A ARR family (Bradi2g61000, Bradi4g43090, Bradi5g25860, Bradi3g45930 and Bradi5g11350) were detected as cytokinin-responsive genes (Fig. 2). In contrast, members of the type B ARR gene family were not detected as cytokinin-responsive genes. These results are consistent with the cytokinin response in Arabidopsis18.

Phylogenic tree of ARR genes and their transcriptional responses to CK in Brachypodium and Arabidopsis.
Brachypodium ARR family members were retrieved using the BLAST software. Similarities in protein sequences (percentage identity) are shown as a phylogenetic tree. Brachypodium genes are shown in red and Arabidopsis genes in black. log2 ratio represents the gene expression ratio between the read count in tZ treatments divided by the average read count from all other treatments. Stat represents the p-value of the microarray experiment in Arabidopsis treated with t-zeatin for 1 h11 or FDR of RNA-seq data when the count from tZ treatment was compared with all other treatments. log2 ratio is hatched in red if genes are up-regulated. cpm represents the average of log2-scaled read counts per million reads from all experiments.
SA response
Eighty-one genes were identified as SA-responsive genes in the high-stringency analysis (Data S3). Genes with the term “defense response to fungus” were enriched in GOE analysis (Table 7). A homolog of the PR-1 gene (Bradi1g57590) was up-regulated by SA treatment (Fig. 3).

Phylogenic tree of PR1, its orthologs and transcriptional responses to SA treatment in Brachypodium and Arabidopsis.
Brachypodium PR1-like genes were retrieved using the BLAST software. Similarities in protein sequences (percentage identity) are shown as a phylogenetic tree. Brachypodium genes are shown in red and Arabidopsis genes in black. log2 ratio represents the gene expression ratio between read counts from SA treatment divided by the average read counts from all other treatments. Stat represents the p-value of the microarray experiment. Arabidopsis treated with SA for 3 h11 for Arabidopsis genes or FDR of RNA-seq data when the counts from SA treatment were compared with all other treatments for Brachypodium genes. log2 ratio is hatched in red if genes are up-regulated and in blue if down-regulated. cpm represents the log2-scaled read count per million reads of RNA-seq data from all treatments.
ABA response
Four hundred and forty-five genes were identified as ABA-responsive genes in the high-stringency analysis (Data S4). Genes with the term “response to abscisic acid stimulus” were enriched in the GOE analysis (Table 7). Genes homologous to those encoding transcription factors involved in transcriptional regulation in response to ABA (i.e., ABF, NAC, Myb, bHLH) were detected as DEGs (Bradi3g00730, Bradi1g59982, Bradi3g50220, Bradi4g32090, Bradi5g17170, Bradi2g22660, Bradi3g57960, Bradi3g19010, Bradi3g38200, Bradi4g29380, Bradi1g22180, Bradi1g11800, Bradi2g41530, Bradi1g20360, Bradi4g35910).
JA response
Three hundred and eighty-three genes were identified as JA-responsive genes in the high-stringency analysis (Data S5). Genes with the term “response to wounding” were enriched in the GOE analysis (Table 7). Genes involved in the biosynthesis of jasmonic acid (LOX3 homolog (Bradi5g11590), LOX4 homolog (Bradi1g72690), AOS homolog (Bradi3g08160, Bradi1g07480, Bradi1g69330), AOC3 homolog (Bradi1g15840), OPR3 homolog (Bradi3g37650) and OPCL1 homolog (Bradi1g76280) were up-regulated by MJ treatment (Fig. 4). The JAZ genes in Arabidopsis encode JA receptors19. Two members of the JAZ family (Bradi3g23190, Bradi1g58490) were also up-regulated in Brachypodium.

Biosynthesis pathway of JA and transcriptional responses in Brachypodium and Arabidopsis.
Homologs of jasmonate biosynthetic genes in Brachypodium were retrieved using the BLAST software. Brachypodium genes are shown in red and Arabidopsis genes in black. Stat represents the p-value of the microarray experiment. Arabidopsis treated by MJ for 3 hours11 and FDR of RNA-seq data when the count of MJ treatment was compared with all other treatments. log2 ratio represents the gene expression ratio between read counts from the MJ treatment divided by the average read counts from all other treatments. log2 ratio is hatched in red if genes are up-regulated. cpm represents log2 scaled read count per million reads of RNA-seq data from all treatments.
GA response
No gene was detected as a DEG in the high-stringency analysis (Data S6). This indicates that the GA-inducible gene expression response is relatively weaker than those to other hormones, which is consistent with the GA response in Arabidopsis11. When the effects on gene expression of the GA biosysthesis inhibitor Phx treatment were compared to those of the mock 3-h and Phx+GA treatments (low-stringency analysis), 224 genes were detected as DEGs (Data S14). Of these genes, those with the term “response to nitrate” were the most enriched in the GOE analysis (Table 8). In Arabidopsis, many GA-biosynthesis genes are also GA-responsive, since there is a negative feedback regulation in the expression of GA-biosynthesis genes11. The read counts of many genes involved in gibberellin biosynthesis (homologs of GA2oxs, GA3oxs and GA20oxs; i.e., Bradi1g56200, Bradi1g56210, Bradi1g56220, Bradi2g16727, Bradi4g23540, Bradi2g16750, Bradi3g49390, Bradi2g32577, Bradi2g24980, Bradi5g16040, Bradi1g59570, Bradi2g06670, Bradi2g57027, Bradi2g34837, Bradi2g19900, Bradi2g50280) were low and none were detected as DEGs (log2 cpm <1, Table S4). Genes of Xyloglucan endotransglucosylase/hydrolases TCH4 (Bradi3g18690) and XTH12 (Bradi3g10310) were significantly up-regulated. Many genes in Peroxidase superfamily (Bradi1g17860, Bradi1g17877, Bradi5g27160, Bradi2g38690, Bradi2g38700, Bradi1g44790, Bradi1g59520, Bradi2g20830, Bradi2g38670, Bradi3g33780, Bradi1g61550, Bradi2g11320, Bradi3g10470, Bradi5g10070, Bradi2g38685, Bradi1g61530, Bradi2g12180, Bradi2g20850, Bradi1g20020, Bradi2g20840, Bradi2g09660, Bradi1g33730, Bradi1g44800, Bradi2g38680, Bradi3g54010) were also up-regulated.
BR response
Only four genes were detected as DEGs in the Brz220 treatment compared with all other treatments (high-stringency analysis, Data S7). This indicates that the BR-inducible gene expression response is relatively weak compared with those to other hormones, which is consistent with the BR response in Arabidopsis11. When the effects on gene expression of the Brz220 treatment were compared with those of the mock 3-h and Brz220+BL treatments (low-stringency analysis), 474 genes were detected as DEGs (Data S15). Of these genes, those with the term “RNA elongation” were enriched in the GOE analysis (Table 8). Genes with the term “photosynthesis” were also enriched in the GOE analysis. P450 genes involved in the biosynthesis of brassinosteroids (Bradi1g15030, homolog of BR6ox2 and Bradi5g12990, homolog of DWF4) were up-regulated by Brz220 compared to the mock 3-h and Brz220+BL treatments (Fig. 5), although the fold change of Bradi5g12990 was less than twice. This is consistent with the BR response in Arabidopsis; BR6ox2 and DWF4 were up-regulated in the det2 mutant of Arabidopsis compared to the wild-type (Fig. 5).

P450 genes involved in BR biosynthesis and transcriptional responses to Brz in Brachypodium and Arabidopsis.
Brachypodium BR biosynthetic genes were retrieved using the BLAST software. Similarities in protein sequences (percentage identity) are shown as a phylogenetic tree. Brachypodium genes are shown in red and Arabidopsis genes in black. log2 ratio represents the gene expression ratio between read counts from the Brz treatment divided by the average read counts from mock 1-h and Brz+BL treatments. Stat represents the p-value of the microarray experiment, Arabidopsis det2 compared with wild-type11 for Arabidopsis genes or FDR of RNA-seq data when the counts from the Brz treatment were compared with mock 1-h and Brz+BL treatments for Brachypodium genes. log2 ratio is hatched in red if genes are up-regulated and in blue if down-regulated. cpm represents log2 scaled read counts per million reads of RNA-seq data from mock 1-h, Brz and Brz+BL treatments.
Ethylene response
No gene was detected as a DEG in response to ethylene in the high-stringency analysis (Data S8). To confirm response of seedlings to ACC treatment under our experimental conditions, plants were treated with ACC for a further week. Seedlings showed growth inhibition when grown in the presence of 10 μM ACC (Fig. S3), indicating that plants respond to exogenous ACC. Therefore, gene expression in response to ACC treatment was compared to that in response to the mock 1-h and mock 3-h treatments (low-stringency analysis). Fifty-two genes were detected as DEGs (Data S16). Of these DEGs, those with the term “RNA elongation” were enriched in GOE analysis (Table 8). A gene homologous to EIN4, which is involved in sensing ethylene (Bradi5g00700) was up-regulated and a gene homologous to ACS6, which is a member of the ethylene synthesis gene family, (Bradi5g19100) was down-regulated by ethylene treatment compared to the mock 1-h and mock 3-h treatments.
Discussion
The comprehensive transcriptional response of Brachypodium to phytohormones was analyzed in this study. To obtain clear responses from Brachypodium, we optimized the experimental design using the expressions of selected Brachypodium marker genes to validate hormone response. The concentrations of phytohormones used in this study were similar (1–10-fold) to those used in the analysis of the response to hormones in rice and Arabidopsis (Table S1), with the exception of the concentration of BL (100-fold compared to that used for Arabidopsis). We successfully identified BR- and GA-regulated genes (e.g., Bradi1g15030, homolog of BR6ox2 as a BR-responsive gene and Bradi3g18690, homolog of TCH4 as a GA-responsive gene).
In rice, 80% of the orthologs of the Brachypodium DEGs were also regulated by CK, ABA, auxin, JA, SA and ethylene. These data show that major transcriptional responses to these phytohormones are shared in monocots, although fewer orthologous genes were commonly regulated by SA and ethylene. It was reported that the SA level in rice was the highest among the tested plants20. This may be one reason for the different responses to SA at similar concentrations. The difference in ethylene-regulated genes between rice and Brachypodium was also reported by Pacheco-Villalobos et al.21. Sato et al.14 also reported the responses of rice seedlings to six hormones: auxin, CK, GA, BR, ABA and JA. However, it was difficult to compare our data with theirs because they reported their data as part of a large transcriptome dataset and no analysis of hormone-responsive gene expression was performed using the data.
The transcriptional responses to phytohormones and the mechanisms involved in transcriptional regulation in Arabidopsis are well characterized. In addition, manually curated GO terms for Arabidopsis is very helpful to analyze large transcriptome dataset. Therefore, we compared the transcriptional response to phytohormones in Brachypodium to that in Arabidopsis thaliana in detail. A comprehensive analysis of phytohormone-regulated genes of Brachypodium revealed common transcriptional responses between Brachypodium and Arabidopsis. To compare the accumulation of GO terms in Brachypodium to Arabidopsis, we assigned new GO terms to the Brachypodium genes using the Arabidopsis GO terms. Some results of the GOE analyses in Arabidopsis and Brachypodium were common following treatment with the same phytohormones. The GO terms “response to auxin stimulus” had the lowest p-values in auxin-regulated DEGs. Similarly, “cellular response to cytokinin stimulus” in CK-regulated DEGs, “defense response to fungus” in SA-regulated DEGs, “response to abscisic acid stimulus” in ABA-regulated DEGs and “response to wounding” in JA-regulated DEGs had the lowest p-values in Brachypodium (Tables 7, 8). These GO terms were also enriched in the hormone-responsive genes in Arabidopsis (Table S3). The GO term “response to nitrate” was enriched in DEGs in the Phx treatment (Table 7), which was consistent with the enriched GO of the GA-biosynthesis mutant ga1-5 when compared to the wild type (Table S5). The GO term “photosynthesis” was enriched in DEGs of Brz-treated Brachypodium (Table 7), which was consistent with the enriched GO terms of the BR-biosynthesis mutant det2 when compared to wild-type Arabidopsis. Consistent with the findings in BR-treated Arabidopsis det2, genes involved in BR-synthesis were detected as DEGs in Brz-treated Brachypodium (Fig. 5). Fewer were detected as DEGs in response to ACC than to other hormones (Tables 4, 5). This was also consistent with transcriptional regulation in ethylene-treated Arabidopsis11,15. These results suggest that many molecular mechanisms characteristic of the responses to phytohormones in Arabidopsis are also present in Brachypodium.
In contrast, there were some differences in the hormone responses of Brachypodium and Arabidopsis. In Arabidopsis, genes involved in ethylene synthesis and signaling, ACO1, ACO3, ACO4/EFE, ACS4, ACS8 and ERF1, are responsive to auxin treatment. Only one gene from the ACO gene family (Bradi2g41840) was detected as a DEG in auxin-treated Brachypodium (Fig. S2). Genes involved in ethylene signaling, such as ERF2, 8, 11, ERS1, EBF2, ETR2, CTR1 and EIN2, are regulated by ACC treatment in Arabidopsis15. However, in Brachypodium, only one gene homologous to EIN4 (Bradi5g00700) was detected as a DEG. These differences in auxin- and ethylene-responsive genes suggest divergent auxin-ethylene relationships between eudicots and monocots. Pacheco-Villalobos et al.21 reported that the cross-talk between ethylene and auxin in Brachypodium differs from that in Arabidopsis. Further analysis of these differentially expressed genes will improve our understanding of the different functions of these hormones and the related genes in Brachypodium and Arabidopsis.
We encountered some difficulties in the analysis of RNA-seq data. We prepared 13–30 million reads because Liu et al.22 reported that the increase in number of DEGs drops after 10–15 million reads. However, the transcriptional responses of some genes might not have been detected because of their low read counts. For example, none of the genes involved in GA-biosynthesis was detected as Phx- or GA-responsive in Brachypodium. However, it was reported that the expressions of the GA-biosynthetic genes AtGA2ox1, AtGA2ox2, GA3ox1 and GA20ox1 changed in Arabidopsis GA-deficient mutants following endogenous GA treatment23,24,25,26. The read counts of these GA-biosynthetic genes in Brachypodium were low (Table S4). In Arabidopsis and rice, few genes respond to ethylene11,13. In this study, no gene in the expansin family was detected as ethylene-responsive in Brachypodium, while AtEXP8 and AtEXP18 in Arabidopsis27 and OsEXP1, OsEXP2 in rice13 are responsive. The expression levels of 27 expansin genes were low (cpm <1, Table S6). The growth of Brachypodium was reduced by ACC treatment (Fig. S3); therefore, some expansin genes may be regulated by ethylene to control cell elongation. To examine the transcriptional regulation of these poorly expressed genes, further analysis using additional RNA-seq reads with different tissues or different growth stages, and/or complementary methods (such as microarray or quantitative PCR) are required.
Methods
Plant materials
Seedlings of Brachypodium distachyon Bd21 were grown on 1/2 MS medium28 supplemented with 1% sucrose and 0.8% agar 4 days after breaking dormancy at 4 °C in the dark. The plants were grown on agar plates for 4 days and transplanted to 1/2 MS liquid culture with 1% sucrose. The plants were pre-incubated for 24 h in liquid culture with shaking (70–80 rpm) and then treated with phytohormones (Table 2). The phytohormones were dissolved in DMSO before addition to the liquid cultures. DMSO (0.1% (v/v)) was used for mock treatments. For the “Inhibitor and GA” and “Inhibitor and BR” treatments, GA and BL were added to liquid cultures 2 h after treatment with the GA inhibitor, prohexadione-calcium (Wako), or BR-inhibitor, brassinazole22029, respectively. All growth and treatment processes were performed at 22 °C under continuous light. Phytohormone treatments were conducted twice independently as biological replicates and the hormone responses were confirmed using the transcriptional responses of marker genes (Table 1). Transcriptional responses of marker genes were checked with RT-PCR using gene specific primers (Table S7) prior to the RNA-seq analysis.
RNA extraction
RNA was extracted from each sample using the RNeasy Mini Kit (QIAGEN). The concentration and quality of the RNA samples were determined using a Nanodrop 2000 instrument (Thermo Scientific). RNA extracted from two biological replicates was mixed and used for the RNA-seq analysis.
Strand-specific RNA-seq analysis
Strand-specific RNA libraries were prepared using the TruSeq Small RNA Sample Prep Kit (Illumina) and TruSeq RNA sample Preparation Kit v2 (Illumina) following the instructions in the Directional mRNA-seq Library Prep. (Pre-Release Protocol Rev.A (Illumina)). Fragmented Poly(A)-RNA was treated with T4 polynucleotide kinase (TAKARA) for phosphorylation of the 5′ end. An RNA 3′ adopter was ligated to the 3′ end using T4 RNA ligase 2, truncated (NEB), then an RNA 5′ adopter was ligated to the 5′ end using T4 RNA ligase 1 (NEB). Single-strand cDNA was synthesized with a primer for the RNA 3′ adaptor. Amplified cDNA was size-selected using 6% polyacrylamide gel electrophoresis, then used as the sequencing library. The quality of all libraries was assessed using a Bioanalyzer 2100 (Agilent). Sequencing was performed using a HiSeq2000 sequencer (Illumina) to sequence 100 bp. The sequencing was performed with the single-read method. The sequence data have been submitted to the DDBJ Sequence Read Archive under accession number PRJDB2997.
Mapping of RNA-Seq reads, transcript assembly and abundance estimation
The sequences were aligned to the Phytozome 9.0 Brachypodium distachyon reference genome Bdistachyon_192 (Bdistachyon_192_hardmasked.fa.gz) using TopHat v2.0.1130, which is integrated with Bowtie v2.2.131. The Bowtie index was generated from Bdistachyon_192 and counting of the mapped reads was performed using Cufflinks v2.2.132 and genome annotation reference PASA_updates.phytozome.gff34. Read counts were generated using Cuffdiff 32. Differentially expressed genes (DEGs) were identified with edgeR16, as follows. Parameters and function usages in edgeR were determined to maximize the specificity for detecting hormone marker genes (Table 1) as DEGs. Read counts were normalized with the function “calcNormFactors”. The TMM normalization method in edgeR can normalize different amounts of RNA-Seq data without increasing the false-positive rate of detecting DEGs33. Dispersions among genes and among replications were estimated by the functions “estimateGLMCommonDisp”, “estimateGLMTrendedDisp” and “estimateGLMTagwiseDisp” and p-values were calculated using the likelihood-ratio test with the generalized linear models. Calculated p-values were adjusted using the false discovery rate (FDR) of Benjamini and Hochberg’s approach34. DEGs were defined as FDR <0.05 and up- or down-regulated genes were identified in the DEGs as those having a log2 ratio of cpm >1 or that of cpm <−1, respectively.
Phytohormone-regulated genes in Arabidopsis
Microarray data from Arabidopsis thaliana treated with phytohormones were retrieved from the AtGenExpress project11. Signals in the microarray data were calculated using R package affy (1.42.3) with the MAS5 method35. DEGs were identified as p-value < 0.1 by Student’s t-test and up- and down-regulated genes were defined in the DEGs as log2 ratios of signals >1 and <−1, respectively. GOE analyses were performed with the DEGs.
Determination of orthologous/homologous genes and generation of the phylogenetic tree
To obtain orthologous gene pairs, Arabidopsis protein sequences (TAIR10) and rice protein sequences (RGAP 7) were applied to BLAST (blastp) to search for homologous sequences in the Brachypodium protein sequence (Bdistachyon_192_peptide.fa from Phytozome 9.0). Brachypodium sequences with top hit were collected and applied to BLAST again to search for the most similar sequences in Arabidopsis and rice. If the results of the second BLAST returned the former Arabidopsis or rice sequence, the corresponding gene pairs were selected as orthologous gene pairs. To obtain genes from each gene family in Brachypodium, protein sequences of members of each family in Arabidopsis were applied to BLAST (blastp with default settings) to search for similar sequences in Brachypodium (Bdistachyon_192_peptide.fa). Brachypodium sequences with E-values less than thresholds were collected and the corresponding Brachypodium genes were selected as family members (Figs 1, 2, 3, 4, 5 and Figs S1,S2). To obtain superfamily or functionally related subfamily, optimal E-value for each gene family was selected. For the Aux/IAA gene family, 19 protein sequences of Arabidopsis Aux/IAA1-19 with a threshold E-value < 10−10 were used. For the ARR family, 17 protein sequences of Arabidopsis ARR1-17 with a threshold E-value < 10−5 were used. For the GH3 gene family, sequences of Arabidopsis genes in Fig. S1 with a threshold E-value < 0.1 were used. For the ACS and ACO gene families, 11 protein sequences (ACS1-11) and 4 proteins (ACO1-4) with a threshold E-value < 10−10 were used. To obtain homologous BR-biosynthetic P450 genes from Brachypodium, the protein sequences of BAS1, Chibi2, BR6ox2, BR6ox1, DWF4, CPD, ROT3 and CYP90D1/At3g13730 were applied to BLAST (blastp) to search for similar sequences in the Brachypodium protein sequence. Brachypodium sequences with an E-value < 10−100 were collected and the corresponding Brachypodium genes were selected as BR-biosynthetic genes. To identify SA-responsive genes homologous to PR1 in Brachypodium, the protein sequence of PR1 and its Arabidopsis homologous proteins (BLAST E-value < 10−10) were applied to BLAST (blastp) to search for similar sequences in the Brachypodium protein sequence. Brachypodium sequences with an E-value < 10−10 were collected and the corresponding Brachypodium genes were selected as PR1-like genes. Similarly, to identify genes homologous to ERF1, the ERF1 protein sequence was applied to BLAST and those with an E-value < 10−10 were selected as ERF1-like genes. To identify genes homologous to JA-biosynthetic genes in Brachypodium, the protein sequences of AtLOX2-4, AOS, AOC3, OPR3, OPCL1, ACX1, ACX5, AIM1, KAT2, JMT and JAR were applied to BLAST (blastp) to search for homologous sequences in the Brachypodium protein sequence. Brachypodium sequences with an E-value < 10−10 were collected and applied to BLAST again to search for the most similar sequences in Arabidopsis. If the results of the second BLAST returned AtLOX2-4, AOS, AOC3, OPR3, OPCL1, ACX1, ACX5, AIM1, KAT2, JMT or JAR, the corresponding Brachypodium genes were selected as being homologous JA-biosynthetic genes. To create the phylogenetic tree of orthologous genes in Brachypodium and Arabidopsis, translated amino acid sequences were aligned with MAFFT36 using the “auto” settings. The neighbor-joining tree37 of the aligned sequences was then generated using percent identity and visualized as a Rectangular Cladogram in Dendroscope38.
Gene ontology term enrichment (GOE) analysis
GOE analysis was carried out using the following procedure. First, Brachypodium protein sequences were applied to BLAST (tblastn) to search for the most similar gene in Arabidopsis thaliana. GO terms of the top hits in Arabidopsis were copied as the customized annotations of corresponding Brachypodium genes. GOE analysis was performed using AgriGO39 with customized annotations as the query, customized annotated reference and with default settings for statistical analysis (p-value was calculated with Fisher’s exact test and “FDR with Yekutieli”).
Additional Information
How to cite this article: Kakei, Y. et al. Transcriptome analysis of hormone-induced gene expression in Brachypodium distachyon. Sci. Rep. 5, 14476; doi: 10.1038/srep14476 (2015).
References
Brkljacic, J. et al. Brachypodium as a Model for the Grasses: Today and the Future. Plant Physiol. 157, 3–13 (2011).
Draper, J. et al. Brachypodium distachyon. A New Model System for Functional Genomics in Grasses. Plant Physiol. 127, 1539–1555 (2001).
International Brachypodium Initiative. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010).
Mochida, K. et al. Large-Scale Collection and Analysis of Full-Length cDNAs from Brachypodium distachyon and Integration with Pooideae Sequence Resources. PLoS ONE 8, e75265 (2013).
Priest, H. D. et al. Analysis of Global Gene Expression in Brachypodium distachyon Reveals Extensive Network Plasticity in Response to Abiotic Stress. PLoS ONE 9, e87499 (2014).
Winter, D. et al. An ‘Electronic Fluorescent Pictograph’ Browser for Exploring and Analyzing Large-Scale Biological Data Sets. PLoS ONE 2, e718 (2007).
Obayashi, T., Nishida, K., Kasahara, K. & Kinoshita, K. ATTED-II Updates: Condition-Specific Gene Coexpression to Extend Coexpression Analyses and Applications to a Broad Range of Flowering Plants. Plant Cell Physiol. 52, 213–219 (2011).
Sasaki, E., Takahashi, C., Asami, T. & Shimada, Y. AtCAST, a Tool for Exploring Gene Expression Similarities among DNA Microarray Experiments Using Networks. Plant Cell Physiol. 52, 169–180 (2011).
Kakei, Y. & Shimada, Y. AtCAST3.0 Update: A Web-Based Tool for Analysis of Transcriptome Data by Searching Similarities in Gene Expression Profiles. Plant Cell Physiol. 56, e7–e7 (2015).
Nagano, A. J. et al. Deciphering and Prediction of Transcriptome Dynamics under Fluctuating Field Conditions. Cell 151, 1358–1369 (2012).
Goda, H. et al. The AtGenExpress hormone and chemical treatment data set: experimental design, data evaluation, model data analysis and data access. Plant J. 55, 526–542 (2008).
Huo, N. et al. Structural characterization of Brachypodium genome and its syntenic relationship with rice and wheat. Plant Mol. Biol. 70, 47–61 (2009).
Garg, R., Tyagi, A. K. & Jain, M. Microarray analysis reveals overlapping and specific transcriptional responses to different plant hormones in rice. Plant Signal Behav. 7, 951–956 (2012).
Sato, Y. et al. RiceXPro Version 3.0: expanding the informatics resource for rice transcriptome. Nucleic Acids Res. 41, D1206–D1213 (2012).
Nemhauser, J. L., Hong, F. & Chory, J. Different Plant Hormones Regulate Similar Processes through Largely Nonoverlapping Transcriptional Responses. Cell 126, 467–475 (2006).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012).
Shimono, M. et al. Rice WRKY45 Plays a Crucial Role in Benzothiadiazole-Inducible Blast Resistance. Plant Cell 19, 2064–2076 (2007).
To, J. P. C. et al. Cytokinin Regulates Type-A Arabidopsis Response Regulator Activity and Protein Stability via Two-Component Phosphorelay. Plant Cell 19, 3901–3914 (2007).
Sheard, L. B. et al. Jasmonate perception by inositol phosphate-potentiated COI1-JAZ co-receptor. Nature 468, 400–405 (2010).
Silverman, P. et al. Salicylic Acid in Rice (Biosynthesis, Conjugation and Possible Role). Plant Physiol. 108, 633–639 (1995).
Pacheco-Villalobos, D., Sankar, M., Ljung, K. & Hardtke, C. S. Disturbed Local Auxin Homeostasis Enhances Cellular Anisotropy and Reveals Alternative Wiring of Auxin-ethylene Crosstalk in Brachypodium distachyon Seminal Roots. PLoS Genet 9, e1003564 (2013).
Liu, Y., Zhou, J. & White, K. P. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics 30, 301–304 (2014).
Chiang, H. H., Hwang, I. & Goodman, H. M. Isolation of the Arabidopsis GA4 locus. Plant Cell Online 7, 195–201 (1995).
Phillips, A. L. et al. Isolation and Expression of Three Gibberellin 20-Oxidase cDNA Clones from Arabidopsis. Plant Physiol. 108, 1049–1057 (1995).
Xu, Y.-L., Li, L., Gage, D. A. & Zeevaart, J. A. D. Feedback Regulation of GA5 Expression and Metabolic Engineering of Gibberellin Levels in Arabidopsis. Plant Cell Online 11, 927–935 (1999).
Thomas, S. G., Phillips, A. L. & Hedden, P. Molecular cloning and functional expression of gibberellin 2- oxidases, multifunctional enzymes involved in gibberellin deactivation. Proc. Natl. Acad. Sci. 96, 4698–4703 (1999).
Cho, H.-T. & Cosgrove, D. J. Regulation of Root Hair Initiation and Expansin Gene Expression in Arabidopsis. Plant Cell Online 14, 3237–3253 (2002).
Murashige, T. & Skoog, F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiol. Plant. 15, 473–497 (1962).
Sekimata, K. et al. A Specific and Potent Inhibitor of Brassinosteroid Biosynthesis Possessing a Dioxolane Ring. J. Agric. Food Chem. 50, 3486–3490 (2002).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53 (2013).
Dillies, M.-A. et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 14, 671–683 (2013).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Li, C., Wong, W. H. & others. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol 2, 1–11 (2001).
Katoh, K. & Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26, 1899–1900 (2010).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425 (1987).
Huson, D. H. & Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. sys062 (2012), 10.1093/sysbio/sys062.
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–70 (2010).
Acknowledgements
We are grateful to Ms. Yuka Mitani, Ms. Ayaka Saito and Dr. Ayako Nakamura for their help in growing plants and examining the expression of marker genes. This paper is contribution No. 1019 from the Kihara Institute for Biological Research, Yokohama City University. This work was supported by JSPS KAKENHI Grant Number 26506016.
Author information
Authors and Affiliations
Contributions
Y.K., K.M., K.S. and Y.S. designed the study. Y.K. performed experiments and drafted the manuscript. Y.K., T.S. and T.Y. performed the statistical analysis of data. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kakei, Y., Mochida, K., Sakurai, T. et al. Transcriptome analysis of hormone-induced gene expression in Brachypodium distachyon. Sci Rep 5, 14476 (2015). https://doi.org/10.1038/srep14476
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14476
This article is cited by
-
Disclosure of salicylic acid and jasmonic acid-responsive genes provides a molecular tool for deciphering stress responses in soybean
Scientific Reports (2021)
-
Genome-wide survey and expression analysis of calcium-dependent protein kinase (CDPK) in grass Brachypodium distachyon
BMC Genomics (2020)
-
Transcriptional reference map of hormone responses in wheat spikes
BMC Genomics (2019)
-
Genome-wide analysis of basic helix-loop-helix (bHLH) transcription factors in Brachypodium distachyon
BMC Genomics (2017)
-
Ethylene-induced transcriptional and hormonal responses at the onset of sugarcane ripening
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
