
Abstract
In cytochrome c oxidase electron transfer from cytochrome c to O2 is linked to transmembrane proton pumping, which contributes to maintaining a proton electrochemical gradient across the membrane. The mechanism by which cytochrome c oxidase couples the exergonic electron transfer to the endergonic proton translocation is not known, but it presumably involves local structural changes that control the alternating proton access to the two sides of the membrane. Such redox-induced structural changes have been observed in X-ray crystallographic studies at residues 423–425 (in the R. sphaeroides oxidase), located near heme a. The aim of the present study is to investigate the functional effects of these structural changes on reaction steps associated with proton pumping. Residue Ser425 was modified using site-directed mutagenesis and time-resolved spectroscopy was used to investigate coupled electron-proton transfer upon reaction of the oxidase with O2. The data indicate that the structural change at position 425 propagates to the D proton pathway, which suggests a link between redox changes at heme a and modulation of intramolecular proton-transfer rates.
Similar content being viewed by others
Introduction
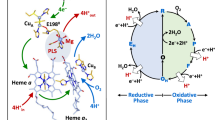
Cytochrome c oxidase (CytcO) is a membrane-bound protein complex, which utilizes the free energy released during electron transfer from cytochrome c to O2, to proton pumping across the membrane. The initial electron acceptor in CytcO is CuA, which is composed of two copper ions. In the aa3-type CytcO from Rhodobacter (R.) sphaeroides electrons are transferred from CuA consecutively to heme a and then to the catalytic site, which is composed of heme a3 and CuB. In this CytcO the proton-pumping stoichiometry is on average about one proton pumped, from the negative (n) side to the positive (p) side of the membrane, per electron transferred to O2. The catalytic site is buried within the membrane-spanning part of the protein, at a distance of ~2/3 of the membrane thickness from the n-side of the membrane. Protons that are involved in reduction of O2 to H2O (O2 + 4H+ + 4e-→2H2O) are transferred through two proton pathways linking the n-side surface with the catalytic site. One of these pathways, denoted by the letter D, is also used for transfer of all protons that are pumped across the membrane. The branching point where protons are directed from the D pathway either to the catalytic site or an acceptor site for the pumped protons (see below) is a highly conserved Glu residue (Glu286) (Fig. 1a), located at a distance of ~25 Å from the n-side surface (for recent publications on the structure and function of the CytcOs, see e.g.1,2,3,4,5,6,7,8,9,10,11,12,13).

Structure and function of CytcO.
(a) The inset shows the overall structure (PDB entry 1M56) of CytcO from R. sphaeroides, the redox-active cofactors involved in electron transfer and the approximate position of the membrane. The main figure shows the relative positions of Ser425, Glu286, hemes a and a3 as well as CuB. The figure was prepared using PyMOL73. (b) The reactions studied in this work (see text for detailed description, the superscripts in the square boxes refer to the number of electrons at the catalytic site).
A redox-driven proton pump must fulfill a number of functional requirements, which are reflected in the structural design of the protein. First, proton pumping across the membrane requires specific proton-conduction pathways spanning the entire distance from the n to the p side surface. However, the proton pathway(s) can not be open to both sides of the membrane simultaneously over time scales of the proton transfer because this arrangement would result in a proton short circuit across the membrane. Consequently, proton transfer must be controlled by the protein. The control element could, for example, be composed of a protonatable residue or a cluster of residues that can rapidly exchange protons with either the n or p side of the membrane. This protonatable site is often referred to as the “proton-loading site”, PLS14,15,16. One possible general design of the proton pump would be to link the alternating proton access to and from the PLS, to intramolecular electron transfer such that the free energy from the electron transfer would be conserved by changing the pKa of the PLS. The pKa of the PLS would be high (relative to the solution pH) when in contact with the proton-input side and low when in contact with the proton-output side.
On the basis of the general considerations outlined above, it is likely that changes in the redox state of the metal cofactors result in local structural changes that control the proton access to the two sides of the membrane. Because the proton-tunneling distance is on the order of ~1 Å, the rate of proton transfer can easily be modulated by minor structural changes17. When considering the aa3 oxidases, early structures of the oxidized and reduced forms of the Paracoccus denitrificans CytcO revealed essentially no structural changes18. Results from more recent amide hydrogen-deuterium exchange Mass Spectrometric studies19,20 and from X-ray crystallographic studies of CytcO from R. sphaeroides and bovine heart indicate that structural changes upon reduction of the oxidized CytcO do occur and that these changes are functionally relevant8,21,22,23,24,25,26 (possible reasons from differences between results from the early and more recent studies are discussed in8).
A segment of the protein that has attracted particular interest is that at helix X, located between the hemes. Here, a significant change in side chain configuration was observed for residue Ser425 (Ser382 in the bovine heart mitochondrial CytcO), ~7 Å from the heme a iron (see Fig. 1a, the change in position is shown in8,22). Similar changes in structure were observed with both the R. sphaeroides8,21,22 and mitochondrial CytcOs23,24. In the mitochondrial oxidase these changes were suggested to be part of the proton pumping mechanism by modulation of proton transfer through the H pathway27. In the R. sphaeroides CytcO the redox-induced changes were suggested to modulate the proton access via the K and D proton pathways, respectively8. Interestingly, the redox induced changes were also linked to changes in water structure, presumably modulating the proton access from Glu286 to the catalytic site. In other words, these results suggest a link between re-allocation of the Ser425 side chain and proton transfer from the D pathway8. These redox-induced structural changes should also modulate proton transfer to the PLS because, as outlined above, an alternating proton access to the PLS must be part of the proton-pumping mechanism.
The distance between Ser425 and Glu286 is ~15 Å. The location of the PLS is not known, but experimental and theoretical data suggest that it is found in a protein segment around one of the His ligands of CuB or the propionic acids of heme a328,29,30,31,32, most likely including a cluster of residues in this region14, all found at a distance of 13–20 Å from Ser425.
On the basis of the discussion above, we reasoned that if the observed structural changes around Ser425 are mechanistically relevant, then a structural modification in the general vicinity of Ser425 should affect proton transfer through the D pathway. Consequently, we used site-directed mutagenesis to replace Ser425 by e.g. Ala and then investigated the kinetics of electron and proton transfer reactions, associated with O2 reduction, in parts of the reaction cycle that are linked to proton pumping via the D pathway. To study these reactions, we measured absorbance changes of the CytcO cofactors as well as that of a pH-sensitive dye in solution after flash photolysis of the reduced CytcO-CO complex in the presence of O2. After dissociation of the CO ligand, O2 binds to heme a3, which is followed in time by step-wise electron transfer from all four redox sites. This approach allows observation of the formation and decay of intermediate states that are populated during reduction of O2 to H2O, catalyzed by CytcO (see Fig. 1b and e.g.33,34,35 for a more detailed description of the technique). The data show that at neutral pH the Ser425Ala structural variant displayed the same electron and proton-transfer rates as the wild-type CytcO. However, at low proton concentrations (pH>9), rapid proton uptake to Glu286 was maintained at high pH, indicating that the structural modification at position 425 resulted in changes of a reaction step that is rate limiting for proton transfer through the D proton pathway.
Results
To investigate the function of the Ser425Ala variant, we first studied internal electron transfer between the hemes in the absence of oxygen36. A state of the CytcO was prepared in which heme a3 and CuB are reduced while heme a and CuA are oxidized and carbon monoxide is bound to heme a3. The CO ligand stabilizes the reduced state of heme a3. Consequently, after light-induced dissociation of CO (Fig. 2, rapid absorbance increase at t = 0) the electron at heme a3 equilibrates with heme a, which is seen as a decrease in absorbance with a time constant of 2.2 ± 0.2 μs (SD of 10 traces) with the wild-type CytcO. As seen in the Figure, this absorbance decrease was faster with the Ser425Ala variant CytcO (τ = 1.0 ± 0.2 μs, SD of 10 traces).

Absorbance changes associated with internal electron transfer in the absence of O2.
A sample of CytcO with heme a/CuA oxidized, heme a3/CuB reduced and CO bound to heme a3 was prepared. The CytcO was illuminated with a short laser flash, which resulted in dissociation of the CO ligand, seen as an increase in absorbance at t = 0. The rapid decrease in absorbance is associated with electron transfer from heme a3 to heme a (τ ≅ 2.2 ± 0.2 μs and 1.0 ± 0.2 μs with the wild-type and Ser425Ala mutant CytcOs, respectively). The absorbance then decreased slowly to reach the original level at t < 0 with time constants of ~65 ms and ~50 ms for the wild-type Ser425Ala CytcOs, respectively. Experimental conditions: the CytcO concentration was ~0.7 μM, 100 mM BIS-TRIS propane, pH 8.5, 0.05% DDM, 0.1 mM EDTA at ~22 °C and ~1.4 mM CO (at 140 kPa). In cases where heme a became fractionally reduced, the enzyme solution was titrated with anaerobic ferricyanide until heme a became re-oxidized as determined from the optical absorption spectra. Ascorbate (2 mM) and the redox mediator hexamine-ruthenium chloride (1 μM) were added to fully reduce the CytcO. The traces have been scaled to 1 μM reacting CytcO.
Next, we studied the reaction of the four-electron reduced CytcO with O2. The CytcO was reduced and incubated under CO atmosphere to form the CytcO-CO complex. The sample was then rapidly mixed with an O2-saturated solution after which the CO ligand was dissociated with a laser flash. The mixing allowed O2 to bind to heme a3 thereby initiating the oxidation of the CytcO. As seen previously (e.g.37), with the wild-type CytcO, at 445 nm (Fig. 3a), the increase in absorbance at t = 0 is associated with dissociation of the CO ligand yielding the reduced CytcO (state R2 in Fig. 1b, the superscript refers to the number of electrons at the catalytic site). It is followed in time by an absorbance decrease with a time constant of ~10 μs associated with binding of O2 (at 1 mM O2) forming state A2. In the next step the electron at heme a is transferred to the catalytic site with a time constant of ~40 μs forming the peroxy state called P3, which is associated with a small decrease in absorbance at 445 nm. This electron transfer is also seen as a decrease in absorbance at 580 nm (Fig. 3b).

Absorbance changes associated with reaction of the reduced CytcO with O2.
At all wavelengths the initial absorbance change at t = 0 is associated with dissociation of the CO ligand. (a) At 445 nm the main contribution is from the heme groups. After CO dissociation, the absorbance decreases as a result of binding of O2 (τ ≅ 10 μs), followed by formation of P3 (τ ≅ 40 μs) and oxidation of the CytcO, i.e. formation of state O4 (τ ≅ 1.0 ms and τ ≅ 1.5 ms for the wild-type and Ser425Ala CytcOs, respectively, at neutral pH). Neutral and high pH are pH 7.0 (WT)/6.5 (S425A) and pH 10 (WT)/10.5 (S425A), respectively. (b) At 580 nm the absorbance decrease in the time range 5 μs–100 μs is associated with formation of the P3 state. The increase in absorbance at neutral pH in the range 100–300 μs is associated with the P3 → F3 reaction with time constants of ~90 μs and ~60 μs for the wild-type and Ser425Ala CytcOs, respectively. The final absorbance decrease is associated with the F3 → O4 reaction. Neutral and high pH are pH 7.4 (WT)/6.5 (S425A) and pH 10 (WT)/10.5 (S425A), respectively. Wild-type data are from43. (c) At 830 nm the main contribution to the absorbance changes is from CuA where an increase in absorbance is associated with oxidation of this redox center. Neutral and high pH are pH 7.4 (WT)/6.5 (S425A) and pH 10.4 (WT)/10.5 (S425A), respectively. Experimental conditions after mixing: CytcO concentration 0.3–1.3 μM, 0.05% DDM, 100 μM EDTA at pH 7.5, 1 mM O2 at ~22 °C. The mixing ratio was 1:5 with oxygen-saturated buffer solutions (100 mM): MES at 6.5; HEPES at pH 7, 7.4 and 7.5; CAPS, pH 10, 10.4 and10.5. The traces have been scaled to 1 μM reacting enzyme.
The electron transfer from heme a to the catalytic site triggers proton uptake from solution (Fig. 4) to the catalytic site, which at neutral pH displays a time constant of ~100 μs at pH 7.4. The proton uptake yields the ferryl state, F, at the catalytic site, which is seen as an increase in absorbance at 580 nm (Fig. 3b). The proton uptake associated with the P3 → F3 reaction is also linked to fractional electron transfer from CuA to heme a, seen as an increase in absorbance at 830 nm with a time constant of ~100 μs (Fig. 3c, oxidation of CuA). In the final step of the reaction, the electron in the CuA—heme a equilibrium is transferred to the catalytic site forming the oxidized CytcO (state O4) with a time constant of ~1 ms at neutral pH. This reaction is seen as a decrease in absorbance at 445 nm and at 580 nm (Fig. 3a,b) and an increase in absorbance at 830 nm (Fig. 3c, oxidation of CuA). A comparison of the traces recorded at neutral pH (Fig. 3) shows only minor differences between the wild-type and Ser425Ala variant CytcOs, except that the P3 → F3 rate was slightly increased from ~1.1·104s−1 to ~1.5·104s−1 (Fig. 5a) and the extent of electron transfer from CuA to heme a (Fig. 3c) was slightly larger with the Ser425Ala CytcO than with the wild-type CytcO, which indicates that at pH ~7 the midpoint potential of heme a was slightly higher with the Ser425Ala than with the wild-type CytcO.

Absorbance changes of the dye phenol red associated with proton uptake during O2 reduction.
The same reaction as in Fig. 3 was studied, but at 560 nm where the main contribution is from the dye. The signal shown is a difference between that obtained without buffer and that obtained with buffer. Experimental conditions after mixing: ~0.6 μM reacting enzyme in 100 mM KCl, pH 7.8, 0.05% DDM, 40 μM phenol red, 100 μM EDTA, 1 mM O2 at ~22 °C. The measurements were then repeated in the presence of 0.1 M HEPES (without KCl) and these absorbance changes were subtracted from those obtained with the un-buffered solution yielding absorbance changes that were only associated with proton uptake. The shown traces are average of 15 experiments.

pH dependence of reaction steps associated with proton uptake.
(a) The P3 → F3 reaction, where the rate constants are derived from absorbance changes at 580 nm and at 830 nm. (b) The F3 → O4 reaction, where the rate constants are derived from absorbance changes at 445 nm, 580 nm and 830 nm. (c) Amplitude of the 830-nm absorbance change (electron transfer CuA —> heme a) in percent of the total change at this wavelength (wild-type data are from3,58).
Results from earlier studies have shown that functional differences caused by minor changes in structure may be revealed at low proton concentrations (e.g.38). Consequently, we investigated the reaction steps that are linked to proton uptake (and pumping) during reaction of the reduced CytcO with O2 also at high pH (Figs 3 and 5). Earlier studies with the wild-type CytcO have shown that the P3 → F3 reaction rate is approximately pH independent up to pH ~9 and then it decreases with increasing pH at higher pH (Fig. 5a). In contrast, with the Ser425Ala variant, the P3 → F3 rate displayed a much weaker pH dependence, the rate constant dropped by less than a factor of ~2 from ~1.3·104 s−1 at pH 6 to ~7.8·103 s−1 at pH 10.5 (Fig. 5a, the rate shown in this figure was obtained by fitting data at both 580 nm (P3 → F3) and at 830 nm (CuA oxidation)). The extent of electron transfer from CuA to heme a was significantly larger with the Ser425Ala than with the wild-type CytcO (Fig. 5c). We also measured the P3 → F3 rate with two other structural variants at the Ser425 position, Ser425Thr and Ser425Val, but only at 3 pH values, i.e. 6, 8 and 10. Qualitatively, the same behavior was observed as with the Ser425Ala variant (see Fig. 5a,b). The F 3→ O4 rate constant displayed similar pH dependence with the Ser425Ala as with the wild-type CytcO.
To investigate whether or not the Ser425Ala variant pumps protons, the CytcO was reconstituted in lipid vesicles with the pH dye phenol red added to the outside of the vesicles (see “Materials and Methods”) and the dye absorbance changes were measured as a function of time during CytcO turnover. The data showed that the Ser425Ala variant pumps protons with about the same stoichiometry as the wild-type CytcO (Fig. 6).

Absorbance changes of the dye phenol red at 558.7 nm associated with proton pumping.
The CytcO was reconstituted in lipid vesicles and the pH dye phenol red was added to the outside of these vesicles to detect changes in proton concentration upon mixing with reduced cytochrome c. Valinomycin was added to deplete the electrical component of the electrochemical gradient (pumping, lower trace). Addition of the proton ionophore, CCCP resulted in total depletion of the electrochemical gradient across the membrane allowing detection of the net consumption of protons during enzymatic turnover (upper trace). Experimental conditions after mixing in a stopped-flow spectrophotometer: 0.25 μM vesicle reconstituted CytcO, 50 μM HEPES, pH 7.4, 45 mM KCl, 44 mM sucrose, 1 mM EDTA, 100 μM phenol red, 8 μM reduced cytochrome c, 2.5 μM valinomycin and 2.5 μM CCCP. The inside of the vesicles contained 0.1 M HEPES at pH 7.4.
Discussion
We have investigated the effect of a structural modification in a segment of CytcO recently shown to be involved in redox-induced structural changes8,21,22,23,24. The activity of the Ser425Ala variant was about the same as that of the wild-type CytcO. Furthermore, at neutral pH internal electron and proton-transfer as well as proton uptake from solution displayed the same rates as with the wild-type CytcO. However, significant differences were observed between the Ser425Ala variant and the wild-type CytcO when the proton supply was limited (i.e. at high pH).
As seen in Fig. 2, after CO dissociation from the mixed-valence CytcO (i.e. CytcO in which heme a3 and CuB are reduced while heme a and CuA are oxidized) a rapid decrease in absorbance at 445 nm was observed. With the R. sphaeroides CytcO, this relatively large absorbance change is associated with electron transfer from heme a3 to heme a36,39, which was faster with the Ser425Ala CytcO (τ ≅ 1.0 μs) than with the wild-type CytcO (τ ≅ 2.2 μs). More recent data suggest that this electron transfer is controlled by a structural relaxation linked to CO dissociation from CuB40, which transiently binds the CO after its dissociation from heme a3. Thus, the data indicate that a structural change around Ser425 modulates the CuB environment. This observation is consistent with the observed changes around Glu286 as a result of the Ser425Ala replacement (see discussion below) because one of the CuB ligands, His284, is separated by only a single residue from Glu286 and a link between changes at Glu286 and at the catalytic site have been observed in the past41.
The data in Fig. 5c show that during the P3 → F3 reaction a significantly larger fraction of heme a becomes reduced with the Ser425Ala (70%) than with the wild-type CytcO (~55%, see also37). This electron is transferred from CuA and the fraction electron transfer reflects the CuA—heme a equilibrium constant, i.e. the difference in midpoint potentials between the two sites. Assuming that the effect of the amino-acid replacement is on heme a and not CuA (because of the much shorter distance from Ser425 to heme a than to CuA), the difference observed in the current study is consistent with an increase in the heme a midpoint potential of ~20 mV.
During reaction of the reduced CytcO with O2, the first step that is associated with proton uptake from the n side (and proton release to the p side of the membrane) is the P3 → F3 reaction42 (τ ≅ 100 μs, see Fig. 4). Because an electron is transferred to the catalytic site during the preceding step, A2 → P3 (τ ≅ 40 μs), the P3 → F3 reaction involves only proton transfer, i.e. there is no internal electron transfer over the time scale of this reaction. As discussed in more detail below, the observed pKa in the pH dependence of the P3 → F3 rate presumably reflects that of Glu286, which is a component of the proton-transfer pathway. Consequently, changes in this pKa value as a result of the Ser425Ala structural modification presumably reflect effects on the Glu286 pKa.
According to a model that we have developed previously to interpret kinetic data from measurements of the P3 → F3 reaction38,43, proton transfer through the D pathway involves a rapid (<100 μs) equilibrium of Glu286 with the n-side solution and the rate-limiting step is proton transfer from Glu286 to the catalytic site. Thus, according to this model the pH dependence of the P3 → F3 rate (kPF) reflects the pKa of Glu286 (pKE286) as outlined in Eq. 1 below:

where αEH is the fraction protonated Glu286 and kH is the proton-transfer rate from Glu286 to the catalytic site. With the wild-type R. sphaeroides CytcO the value of pKE286 was found to be 9.443,44. In the following sections we argue that the pKa of Glu286 in the Ser425Ala variant is higher than that in the wild-type CytcO.
As already indicated above, the Glu286 residue is located at the end of the D pathway from where protons are transferred either to the catalytic site or toward the PLS. The proton flux to the two sites is presumably modulated by the Glu286 side chain conformation10. Furthermore, the Glu286 connectivity to the catalytic site could also be regulated by redox-induced reallocation of water molecules between the two sites21. The model in Eq. 1 is a simplistic description of the problem because in the P3 → F3 reaction two protons are transferred simultaneously via Glu286, one to the PLS and one to the catalytic site. The mechanism by which the Glu286 controls the proton flux to the two acceptors is not understood45,46, but on the basis of results from earlier studies8,10,47,48,49,50,51,52,53 we speculate on the involvement of Glu286. It is likely that during each of the four reactions steps that are linked to proton pumping (such as e.g. the two reaction steps, P3 → F3 and F3 → O4, studied here) the Glu side chain would switch from its relaxed configuration, where it is in contact with the n-side of the membrane, to a transient conformation from where it could transfer a proton to the PLS or the catalytic site. According to a previously presented model, the pKas are different in these two configurations44,51,54,55. The changes in configuration and pKa would be repeated four times during each reaction cycle, i.e. for each transition that is linked to proton pumping. According to the model44,51,54,55 the regulatory function of Glu286 was included by assuming two conformations of the Glu286 side chain, in rapid equilibrium (≪100 μs), with two different pKa values:

where E1 and E2 are the two conformations of Glu286 (Ex− and ExH, deprotonated and protonated states, respectively) and pKa1 and pKa2 are the pKa values of Glu286 in these two states. K1 and K2 are equilibrium constants and pT indicates proton transfer from state E2H to the catalytic site (CS). The apparent (observed) pKa value of Glu286, pKapp, (defined as the one half of the maximum [E2H] value) is:

Here, in the E1 configuration the side chain would be in contact with the D pathway, i.e. the n-side of the membrane. In E2 the side chain would be in contact with the catalytic site and presumably also with the PLS although the latter is not explicitly included in the model in Scheme 1 (discussed in more detail below). According to this model the observed pKa value in measurements of e.g. the P3 → F3 kinetics also reports the equilibrium constant between the two Glu286 side chain configurations44,51,55. It should be noted that the model outlined in Scheme 1 qualitatively describes the scenario in each of the four reaction steps that are linked to proton pumping (four consecutive electron transfers from cytochrome c to O2 occur for each turnover). However, the actual values of the parameters may vary slightly between the four steps, e.g. due to differences in the overall charge distribution within the protein and the presence of an electrochemical potential across the membrane.
Results from earlier functional studies of a large number of structural variants of aa3 CytcOs showed that a number of specific single or double amino-acid residue replacements within the entire span of the D pathway result in CytcO that is active (i.e. reduces O2 to H2O), but in which proton pumping is completely blocked38,55,56,57,58,59,60,61. Detailed kinetic studies of specific electron- and proton-transfer reactions in these CytcO variants revealed an altered pH dependence of the P3 → F3 reaction, interpreted in terms of an altered apparent pKa of Glu28644,51. We suggested that the molecular explanation for this pKapp shift was a change in the equilibrium constant between the two Glu286 side-chain conformations (see Scheme 1)44,51. We note that a change in the K1 and K2 values in Scheme 1. a change in equilibrium between the two states) would result in a change in pKapp even if pKa1 and pKa2 were unaltered. Furthermore, a structural alteration that changes the equilibrium constant between the two Glu286 positions would most likely also alter the relative rates of reactions that follow in time after the E1 to E2 switch. When this switch to the E2 configuration has occurred there are three competing reactions: (i) proton transfer to the catalytic site, (ii) proton transfer to the PLS and (iii) the structural relaxation from state E2 back to E1. As discussed earlier51,54, proton pumping requires that either the proton to PLS is transferred first or, if proton transfer to the catalytic site occurs first, the free energy of this reaction is conserved in a local structural change that drives a consecutive protonation of the PLS. The model outlined in Scheme 1 is a minimum to describe the pH dependence of e.g. the P3 → F3 reaction and, as already mentioned above, it does not explicitly include proton transfer to the PLS because this proton transfer is not studied in the experiments that the model is based on. Nevertheless, protonation of the PLS requires that the relative rates of the three reactions (i)—(iii) above fulfill the requirement that PLS becomes protonated before the structural relaxation takes place. Proton pumping can tolerate a change in all three rates constants provided that their relative values are such that PLS receives a proton. If this requirement is not fulfilled, which is suggested for the uncoupled mutants discussed above, PLS does not receive a proton.
In the context of the above discussion we also note that the change in observed Glu286 pKa and absence of proton pumping would both be result of structural changes around the Glu286 site. However, these two effects must not necessarily be interdependent as the pKa reflects the equilibrium constant between conformations 1 and 2 in Scheme 1, while the proton-pumping efficiency would be determined by the relative rates of events (i)-(iii). Several other models explaining the lack of proton pumping in e.g. the Asn139Asp variant CytcO have been presented (see e.g.51). Here, we focus on the model outlined in Scheme 1 above.
As seen in Fig. 5, also with the Ser425Ala variant the P3 → F3 reaction rate was essentially pH independent in the measured pH range, i.e. the pKa associated with this reaction was shifted to a value >10.5. In the context of the model outlined above, the structural change as a result of the Ser425Ala replacement may alter the Glu286 environment to modify the equilibrium constant between positions 1 and 2, resulting in an increased observed pKa. To explain that the Ser425Ala variant still pumps protons we speculate that in the mutant the ratio of the proton-transfer rates from Glu286 to the catalytic site, to the PLS and the structural relaxation ((i)-(iii) above) would be such that the stoichiometry of the proton pump would not be decreased (c.f. the Ser425Ala variant pumps protons). It should also be noted that the experiments described in this study were carried out in the absence of any electrochemical potential across the membrane. When present, this potential would presumably act to decrease proton transfer from Glu286 to the PLS more significantly than from Glu286 to the catalytic site because the former has a larger component perpendicular to the membrane. Consequently, it is possible that the Ser425Ala variant would display a smaller proton-pumping stoichiometry than the wild-type CytcO in the living cell. A variable proton pumping stoichiometry depending on the proton electrochemical gradient has been addressed in a recent theoretical study12.
Wikström and Verkhovsky suggested an alternative model to explain the decrease in the P3 → F3 rate at pH>9 with the wild-type CytcO. They assumed that proton uptake into the D pathway occurs via a proton “antenna”62 surrounding the D-pathway orifice and that the slowed proton uptake at high pH is due to exhaustion of protons from these groups. Furthermore, they explained the absence of the pH-dependence in the P3 → F3 rate with e.g. the Asn139Asp variant in terms of an altered equilibrium between different configurations of the Glu286 side chain63, in part based on the observation of differences in the structure of the structural variant25 and the wild-type CytcO. Because it is unlikely that the Ser425Ala replacement would result in changes at the D pathway orifice (because of the large distance), also the “alternative view” would explain the altered pH dependence (of the P3 → F3 reaction) in the Ser425Ala variant in terms of changes of the Glu286 configuration.
The discussion above is focused on the P3 → F3 reaction because this step is only associated with proton transfer through the D pathway and there is no interference from internal electron transfer. Also the F3 → O4 reaction involves proton transfer through the D pathway. However, this reaction is also linked to simultaneous electron transfer to the catalytic site, which complicates a detailed quantitative interpretation of the data because both electron and proton transfer determine the overall F3 → O4 rate64 (see also65).
To summarize, the structural data obtained earlier with both the mitochondrial and the R. sphaeroides CytcOs show significant structural changes of Ser425 and surrounding residues upon redox changes of heme a8,21,22,23,24. The data from the present study show that the Ser425Ala replacement results in altering specific reaction steps that are linked to proton pumping. In other words, the combined structural data and the functional data from this study point to a link between redox reactions and proton pumping. Our findings are also consistent with earlier studies using Fourier Transform Infrared Spectroscopy (FTIR) showing a link between redox and structural changes at Glu28666,67. The above-discussed model explains qualitatively these effects in terms of a link between changes at Ser425 and Glu286. However, the discussed changes at Glu286 are likely to comprise only part of the problem to describe the mechanism of proton pumping. As already noted above, the D pathway in the bacterial CytcO is used to transfer both the proton that is used in the reaction that drives the pump and to transfer the proton that is pumped across the membrane. This scenario requires control of the proton flux at two levels: (a) at Glu286 to control the timing of proton transfer to the catalytic site and the PLS, respectively, (b) at the PLS where the flux of the pumped protons across the membrane must be controlled (c.f. proton gating). Problem (a) requires control of the proton flux via Glu286, where the two proton-transfer trajectories branch47, while problem (b) requires control of the proton access to and from the PLS toward the p side.
Even though the above-described model (Scheme 1 and Equations 1, 2) is focused only on problem (a), proton gating at Glu286 and at the PLS are presumably tightly coupled. As briefly outlined in the Introduction section, results from theoretical studies indicate that the PLS is located around the heme a3 propionic acids and most likely involves also a cluster surrounding residues14. Structural changes in this protein segment, around propionate A of heme a3, have been suggested to take place upon reduction of heme a. This coupling may be important both for controlling protonation of the PLS and it’s proton connectivity to the two sides of the membrane31. Taken together, the earlier observations combined with those from the current study point to a link between redox changes at the cofactors, structural changes at Ser425, structural changes at Glu286 as well as at the putative PLS at the heme a3 propionic acids.
Materials and Methods
Site-directed mutagenesis, protein expression and purification
The Ser425 to Ala/Val/Thr mutations were introduced into the CytcO SUI-containing plasmid pJS3-SH, using the Quik-Change site-directed mutagenesis kit (Stratagene/Agilent technologies) as described in68. The amino-acid replacements were verified by sequencing. A DNA fragment containing the mutated site was introduced into the plasmid pRK-415, which holds SU I-III of R. sphaeroides CytcO. The pRK-415 plasmid was transformed into E. coli strain S-17 by electroporation and thereafter transferred to the R. sphaeroides strain JS100 by conjugation. For CytcO expression, R. sphaeroides cells were grown aerobically in shaker incubators at 30 °C in the dark. The inner membrane fraction was collected by ultracentrifugation and CytcO was solubilized in n-dodecyl β-D-maltoside (DDM). The CytcO (His-tagged) was purified using Ni2+-NTA affinity chromatography, essentially as described in68,69.
Steady-state activity
A buffer composed of 50 mM K-Pi (pH 6.7), 0.1% DDM and 1.0 mg/ml asolectin, supplemented with 6 mM ascorbate, 670 μM N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD, used as an electron mediator) and 32 μM cytochrome c was added to the reaction chamber of a Clark-type O2 electrode (Hansatech instruments). CytcO at a concentration of 1 μM in a buffer containing 100 mM Hepes (pH 7.5) and 0.05% DDM was added to the reaction chamber to a final CytcO concentration of 7 nM. The oxygen-consumption rate during CytcO turnover was monitored. The steady-state activities of Ser425Ala, Ser425Val and Ser425Thr CytcOs were 70%, 19% and 16% of that of the wild-type CytcO, respectively (~570 e−/s/CytcO).
Measurement of the oxidation kinetics
Cytochrome c oxidase was diluted to ~10 μM in a buffer composed of 0.1 M HEPES (pH 7.5) and 0.05% DDM and transferred to a locally manufactured Thunberg cuvette. The air in the cuvette was evacuated on a vacuum line and replaced by N2. The CytcO was reduced upon addition of ascorbate (2 mM) and hexaammin-rutheniumchloride (1 μM). The redox state of the enzyme was monitored spectrophotometrically and when full reduction was reached, the N2 atmosphere in the cuvette was exchanged for carbon monoxide (CO). CO binds to reduced heme a3, forming a CO-CytcO complex.
The CO-CytcO complex was mixed in about 10 ms (1:5, CytcO:O2-saturated solution) with an oxygen-saturated buffer (0.1 M HEPES (pH 7.5), 0.05% DDM, 0.1 mM EDTA) in a flow-flash apparatus (Applied Photophysics). About 200 ms after mixing, the CO ligand was dissociated from heme a3 by means of a 10 ns laser flash (Quantel Brilliant B, Nd-YAG, 532 nm), enabling oxygen to bind to heme a3. Absorbance changes associated with the reaction of the reduced CytcO with O2 were monitored as a function of time in a 10-mm cuvette (see also70).
The pH dependence of the reaction of the reduced CytcO with O2 was studied as follows43. The CytcO samples were diluted in 1 mM HEPES, 0.05% DDM, 100 μM EDTA at pH 7.5. The CO-CytcO complex was then rapidly mixed (1:5) with oxygen-saturated buffer solutions (100 mM), supplemented with 0.05% DDM, 0.1 mM EDTA, at different pH values: MES at pH 6 and 6.5; HEPES at pH 7, 7.5 and 8; BIS-TRIS propane, pH 7–9; CHES, pH 9 and 9.5; CAPS, pH 10, 10.5 and 11.
Proton uptake measurements
Two PD-10 columns were used to exchange the buffer of the CytcO samples to 0.1 M KCl, pH 7.8, 0.05% DDM, 0.1 mM EDTA and the CytcO concentration was adjusted to 10 μM. The samples were then treated as described above in the section “ Measurement of the oxidation kinetics” with the exception that the concentration of hexaammin-ruthenium chloride was 0.2 μM. The oxygen-saturated solution contained 0.1 M KCl, pH 7.8, 0.05% DDM, 0.1 mM EDTA and 40 μM phenol red. Typically, 15 traces at 560 nm were collected and averaged. The measurements were then repeated in the presence of 0.1 M HEPES, pH 7.8, 0.05% DDM, 0.1 mM EDTA and these absorbance changes were subtracted from those obtained with the un-buffered solution yielding absorbance changes that were only associated with changes in the proton concentration. The exhaust from the mixing device (unbuffered solution) was collected under N2 atmosphere and then titrated with well-defined amounts of HCl to calibrate the absorbance changes to changes in proton concentration.
Preparation of mixed valence CO-CytcO and flash photolysis measurements
First, the CytcO sample buffer was exchanged to 0.1 M BIS-TRIS propane, pH 8.5, 0.05% DDM, 0.1 mM EDTA on a PD-10 column and the CytcO sample at a concentration of 10 μM was transferred to a modified Thunberg cuvette. The atmosphere in the cuvette was exchanged for N2 and then to CO. During incubation of CytcO under pure CO atmosphere the catalytic site (heme a3/CuB) becomes reduced and CO binds to the reduced heme a3. The CytcO with oxidized CuA/heme a and reduced heme a3/CuB is referred to as the “mixed-valance state”. The CO ligand was dissociated by applying a 10 ns laser flash (Quantel Brilliant Nd:YAG, 532 nm) in a flash photolysis setup (Applied Photophysics), allowing electron transfer to be monitored at different single wavelengths. To improve the time resolution from μs to ns, the measuring light was pulsed (~1 ms pulses, which allowed measuring absorbance changes over ~500–600 μs).
Reconstitution of cytochrome c oxidase in vesicles and proton pumping measurements
A lipid extract was made from soybean phosphatidylcholine (type II, Sigma) using diethyl ether followed by precipitation in acetone. The lipids were dried under a stream of N2. Small unilamellar vesicles (SUVs) were prepared by sonication of 40 mg/ml of the purified lipid extract in 0.1 M HEPES- at pH 7.4 with 2% sodium cholate, using a tip sonicator (Ultrasonic VCX 130, Chemical Instruments AB) at 40% energy output during 30 s on and 30 s off repetitive cycles for a total of 2 min/ml. After sonication, the vesicle solution was centrifuged at 1500 g for 20 min to remove titanium particles and lipid aggregates. Reconstitution of CytcO in the vesicles was performed using a modified version of the protocol described in42,71. Briefly, the purified CytcO was diluted to 6 μM in 0.1 M HEPES at pH 7.4 with 4% sodium cholate and was mixed with the lipid solution at a ratio of 1:1. Detergent was removed using Bio-Beads SM-2 Adsorbent (Bio-Rad Laboratories) allowing the CytcO to insert into the vesicles. The buffer was exchanged for 100 mM KCl, pH 7.4 using a PD-10 column. The CytcO turnover activity was measured as described above. This activity diminishes as an electrochemical proton gradient is built up across the membrane. Upon addition of ionophores (valinomycin and carbonyl cyanide 3-chlorophenylhydrazone (CCCP)) the gradient is removed and the activity increases again. The ratio of the rates obtained after and before addition of the ionophores is the respiratory-control ratio (RCR). Values in the range 4–7 were obtained.
The proton pumping measurements were performed as described in72. Briefly, liposome-reconstituted CytcO was diluted to 0.5 μM CytcO in 50 μM HEPES, pH 7.6, 45 mM KCl, 44 mM sucrose, 1 mM EDTA, 100 μM phenol red and mixed rapidly at a 1:1 ratio with 16 μM reduced cytochrome c in the same solution as the reconstituted enzyme in a stopped-flow spectrophotometer (Applied Photophysics). Absorbance changes of the pH sensitive dye were detected at 558,7 nm (the isosbestic point for cytochrome c oxidation). Valinomycin was added to the reconstituted vesicles at a concentration of 5 μM. In order to detect net consumption of protons during enzymatic turnover (for calibration purposes), the proton ionophore CCCP at 5 μM was finally added to the CytcO-vesicle solution.
Additional Information
How to cite this article: Vilhjálmsdóttir, J. et al. Structural Changes and Proton Transfer in Cytochrome c Oxidase. Sci. Rep. 5, 12047; doi: 10.1038/srep12047 (2015).
References
Hosler, J. P., Ferguson-Miller, S. & Mills, D. A. Energy transduction: Proton transfer through the respiratory complexes. Annual Review of Biochemistry 75, 165–187 (2006).
Yoshikawa, S. et al. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochimica et Biophysica Acta–Bioenergetics 1757, 1110–1116 (2006).
Namslauer, A. & Brzezinski, P. Structural elements involved in electron-coupled proton transfer in cytochrome c oxidase. FEBS Lett 567, 103–110 (2004).
Brzezinski, P. & Gennis, R. B. Cytochrome c oxidase: exciting progress and remaining mysteries. J Bioenerg Biomembr 40, 521–531 (2008).
Brzezinski, P. & Ädelroth, P. Design principles of proton-pumping haem-copper oxidases. Curr Opin Struct Biol 16, 465–472 (2006).
Richter, O. M. H. & Ludwig, B. Electron transfer and energy transduction in the terminal part of the respiratory chain–Lessons from bacterial model systems. Biochimica et Biophysica Acta–Bioenergetics 1787, 626–634 (2009).
Belevich, I. & Verkhovsky, M. I. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxid Redox Signal 10, 1–29 (2008).
Ferguson-Miller, S., Hiser, C. & Liu, J. Gating and regulation of the cytochrome c oxidase proton pump. Biochimica et Biophysica Acta 1817, 489–494 (2012).
Rich, P. R. & Maréchal, A. Functions of the hydrophilic channels in protonmotive cytochrome c oxidase. Journal of the Royal Society Interface 10, 183–196 (2013).
Kaila, V. R. I., Verkhovsky, M. I. & Wikström, M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev 110, 7062–7081 (2010).
Konstantinov, A. A. . Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett 586, 630–639 (2012).
Blomberg, M. R. A., Siegbahn, P. E. M. Proton pumping in Cytochrome c oxidase: Energetic requirements and the role of two proton channels. Biochimica et Biophysica Acta 1837, 1165–1177 (2014).
Popović, D. M., Leontyev, I. V., Beech, D. G. & Stuchebrukhov, A. A. Similarity of cytochrome c oxidases in different organisms. Proteins: Structure, Function and Bioinformatics 78, 2691–2698 (2010).
Lu, J. & Gunner, M. R. Characterizing the proton loading site in cytochrome c oxidase. Proceedings of the National Academy of Sciences 111, 12414–12419 (2014).
Popović, D. M. & Stuchebrukhov, A. A. Electrostatic study of the proton pumping mechanism in bovine heart cytochrome c oxidase. J Am Chem Soc 126, 1858–1871 (2004).
Seibold, S. A., Mills, D. A., Ferguson-Miller, S. & Cukier, R. I. Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: A molecular dynamics comparison of the wild type and R481K mutant. Biochemistry 44, 10475–10485 (2005).
Wraight, C. A. Chance and design-Proton transfer in water, channels and bioenergetic proteins. Biochimica et Biophysica Acta–Bioenergetics 1757, 886–912 (2006).
Harrenga, A. & Michel, H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J Biol Chem 274, 33296–33299 (1999).
Busenlehner, L. S., Brändén, G., Namslauer, I., Brzezinski, P. & Armstrong, R. N. Structural elements involved in proton translocation by cytochrome c oxidase as revealed by backbone amide hydrogen-deuterium exchange of the E286H mutant. Biochemistry 47, 73–83 (2008).
Busenlehner, L. S., Salomonsson, L., Brzezinski, P. & Armstrong, R. N. Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc Natl Acad Sci USA 103, 15398–15403 (2006).
Qin, L., Liu, J., Mills, D. A., Proshlyakov, D. A., Hiser, C. & Ferguson-Miller, S. Redox-dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry 48, 5121–5130 (2009).
Liu, J., Qin, L. & Ferguson-Miller, S. Crystallographic and online spectral evidence for role of conformational change and conserved water in cytochrome oxidase proton pump. Proc Natl Acad Sci USA 108, 1284–1289 (2011).
Muramoto, K. et al. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc Natl Acad Sci USA 107, 7740–7745 (2010).
Tsukihara, T. et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci USA 100, 15304–15309 (2003).
Durr, K. L. et al. A D-pathway mutation decouples the Paracoccus denitrificans cytochrome c oxidase by altering the side-chain orientation of a distant conserved glutamate. J Mol Biol 384, 865–877 (2008).
Svensson-Ek, M., Abramson, J., Larsson, G., Törnroth, S., Brzezinski, P. & Iwata, S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol 321, 329–339 (2002).
Yoshikawa, S., Muramoto, K. & Shinzawa-Itoh, K. Proton-pumping mechanism of cytochrome c oxidase. Annu Rev Biophys Biomolec Struct 40, 205–223 (2011).
Sugitani, R., Medvedev, E. S. & Stuchebrukhov, A. A. Theoretical and computational analysis of the membrane potential generated by cytochrome c oxidase upon single electron injection into the enzyme. Biochimica et Biophysica Acta–Bioenergetics 1777, 1129–1139 (2008).
Goyal, P., Lu, J., Yang, S., Gunner, M. R. & Cui, Q. Changing hydration level in an internal cavity modulates the proton affinity of a key glutamate in cytochrome c oxidase. Proc Natl Acad Sci USA 110, 18886–18891 (2013).
Belevich, I., Bloch, D. A., Belevich, N., Wikström, M. & Verkhovsky, M. I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci USA 104, 2685–2690 (2007).
Kaila, V. R. I., Sharma, V. & Wikström, M. The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochimica et Biophysica Acta–Bioenergetics 1807, 80–84 (2011).
Quenneville, J., Popovic, D. M. & Stuchebrukhov, A. A. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim Biophys Acta 1757, 1035–1046 (2006).
Von Ballmoos, C., Gennis, R. B., Ädelroth, P. & Brzezinski, P. Kinetic design of the respiratory oxidases. Proc Natl Acad Sci USA 108, 11057–11062 (2011).
Brzezinski, P., Öjemyr, L. N. & Ädelroth, P. Intermediates generated during the reaction of reduced Rhodobacter sphaeroides cytochrome c oxidase with dioxygen. Biochimica et Biophysica Acta–Bioenergetics 1827, 843–847 (2013).
Näsvik, Öjemyr L, Maréchal, A., Vestin, H., Meunier, B., Rich, P. R. & Brzezinski, P. Reaction of wild-type and Glu243Asp variant yeast cytochrome c oxidase with O2. Biochimica et Biophysica Actaz–Bioenergetics 1837, 1012–1018 (2014).
Ädelroth, P., Brzezinski, P. & Malmström, B. G. Internal electron transfer in cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry 34, 2844–2849 (1995).
Ädelroth, P., Ek, M. & Brzezinski, P. Factors Determining Electron-Transfer Rates in Cytochrome c Oxidase: Investigation of the Oxygen Reaction in the R. sphaeroides and Bovine Enzymes. Biochim Biophys Acta 1367, 107–117 (1998).
Namslauer, A., Pawate, A. S., Gennis, R. B. & Brzezinski, P. Redox-coupled proton translocation in biological systems: Proton shuttling in cytochrome c oxidase. Proc Natl Acad Sci USA 100, 15543–15547 (2003).
Brzezinski, P. Internal electron-transfer reactions in cytochrome c oxidase. Biochemistry 35, 5612–5615 (1996).
Pilet, E., Jasaitis, A., Liebl, U. & Vos, M. H. Electron transfer between hemes in mammalian cytochrome c oxidase. Proc Natl Acad Sci USA 101, 16198–16203 (2004).
Heitbrink, D., Sigurdson, H., Bolwien, C., Brzezinski, P. & Heberle, J. Transient binding of CO to Cu-B in cytochrome c oxidase is dynamically linked to structural changes around a carboxyl group: A time-resolved step-scan Fourier transform infrared investigation. Biophys J 82, 1–10 (2002).
Faxén, K., Gilderson, G., Ädelroth, P. & Brzezinski, P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature 437, 286–289 (2005).
Namslauer, A., Aagaard, A., Katsonouri, A. & Brzezinski, P. Intramolecular proton-transfer reactions in a membrane-bound proton pump: the effect of pH on the peroxy to ferryl transition in cytochrome c oxidase. Biochemsitry 42, 1488–1498 (2003).
Chakrabarty, S., Namslauer, I., Brzezinski, P. & Warshel, A. Exploration of the cytochrome c oxidase pathway puzzle and examination of the origin of elusive mutational effects. Biochimica et Biophysica Acta–Bioenergetics 1807, 413–426 (2011).
Gunner, M. R., Amin, M., Zhu, X. & Lu, J. Molecular mechanisms for generating transmembrane proton gradients. Biochimica et Biophysica Acta–Bioenergetics 1827, 892–913 (2013).
Yang, S. & Cui, Q. Glu-286 rotation and water wire reorientation are unlikely the gating elements for proton pumping in cytochrome c oxidase. Biophys J 101, 61–69 (2011).
Kaila, V. R. I., Verkhovsky, M. I., Hummer, G. & Wikström, M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc Natl Acad Sci USA 105, 6255–6259 (2008).
Pisliakov, A. V., Sharma, P. K., Chu, Z. T., Haranczyk, M. & Warshel, A. Electrostatic basis for the unidirectionality of the primary proton transfer in cytochrome c oxidase. Proc Natl Acad Sci USA 105, 7726–7731 (2008).
Popović, D. M. & Stuchebrukhov, A. A. Coupled electron and proton transfer reactions during the O→e transition in bovine cytochrome c oxidase. Biochimica et Biophysica Acta–Bioenergetics 1817, 506–517 (2012).
Popovic, D. M. & Stuchebrukhov, A. A. Two conformational states of Glu242 and pKas in bovine cytochrome c oxidase. Photochemical and Photobiological Sciences 5, 611–620 (2006).
Brzezinski, P. & Johansson, A. L. Variable proton-pumping stoichiometry in structural variants of cytochrome c oxidase. Biochimica et Biophysica Acta–Bioenergetics 1797, 710–723 (2010).
Pomès, R., Hummer, G. & Wikström, M. Structure and dynamics of a proton shuttle in cytochrome c oxidase. Biochim Biophys Acta 1365, 255–260 (1998).
Fadda, E., Yu, C. H. & Pomès, R. Electrostatic control of proton pumping in cytochrome c oxidase. Biochimica et Biophysica Acta–Bioenergetics 1777, 277–284 (2008).
Brzezinski, P. & Larsson, G. Redox-driven proton pumping by heme-copper oxidases. Biochim Biophys Acta 1605, 1–13 (2003).
Johansson, A. L., Chakrabarty, S., Berthold, C. L., Högbom, M., Warshel, A. & Brzezinski, P. Proton-transport mechanisms in cytochrome c oxidase revealed by studies of kinetic isotope effects. Biochimica et Biophysica Acta–Bioenergetics 1807, 1083–1094 (2011).
Pfitzner, U., et al. Tracing the D-pathway in reconstituted site-directed mutants of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry 39, 6756–6762 (2000).
Zhu, J., Han, H., Pawate, A. & Gennis, R. B. Decoupling mutations in the D-Channel of the aa3-Type cytochrome c oxidase from Rhodobacter sphaeroides suggest that a continuous hydrogen-bonded chain of waters is essential for proton pumping. Biochemistry 49, 4476–4482 (2010).
Lepp, H., Salomonsson, L., Zhu, J. P., Gennis, R. B. & Brzezinski, P. Impaired proton pumping in cytochrome c oxidase upon structural alteration of the D pathway. Biochim Biophys Acta 1777, 897–903 (2008).
Brändén, G., Pawate, A. S., Gennis, R. B. & Brzezinski, P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci USA 103, 317–322 (2006).
Siletsky, S. A., Pawate, A. S., Weiss, K., Gennis, R. B. & Konstantinov, A. A. Transmembrane charge separation during the ferryl-oxo -> oxidized transition in a nonpumping mutant of cytochrome c oxidase. J Biol Chem 279, 52558–52565 (2004).
Han, D. et al. Replacing Asn207 by Aspartate at the Neck of the D Channel in the aa3-Type Cytochrome c Oxidase from Rhodobacter sphaeroides Results in Decoupling the Proton Pump. Biochemistry 45, 14064–14074 (2006).
Gutman, M. & Nachliel, E. Time-resolved dynamics of proton transfer in proteinous systems. Annu Rev Phys Chem 48, 329–356 (1997).
Wikström, M. & Verkhovsky, M. I. The D-channel of cytochrome oxidase: An alternative view. Biochimica et Biophysica Acta–Bioenergetics 1807, 1273–1278 (2011).
Brändén, G., Brändén, M., Schmidt, B., Mills, D. A., Ferguson-Miller, S. & Brzezinski, P. The protonation state of a heme propionate controls electron transfer in cytochrome c oxidase. Biochemistry 44, 10466–10474 (2005).
Karpefors, M., Ädelroth, P., Zhen, Y., Ferguson-Miller, S. & Brzezinski, P. Proton uptake controls electron transfer in cytochrome c oxidase. Proc Natl Acad Sci USA 95, 13606–13611 (1998).
Hellwig, P. et al. Involvement of glutamic acid 278 in the redox reaction of the cytochrome c oxidase from Paracoccus denitrificans investigated by FTIR spectroscopy. Biochemistry 37, 7390–7399 (1998).
Lübben, M., Prutsch, A., Mamat, B. & Gerwert, K. Electron transfer induces side-chain conformational changes of glutamate-286 from cytochrome bo3. Biochemistry 38, 2048–2056 (1999).
Mitchell, D. M. & Gennis, R. B. Rapid purification of wildtype and mutant cytochrome c oxidase from Rhodobacter sphaeroides by Ni(2+)-NTA affinity chromatography. FEBS Lett 368, 148–150 (1995).
Zhen, Y. et al. Overexpression and purification of cytochrome coxidase from Rhodobacter sphaeroides. Protein Expression and Purification. Protein Expr Purif 13, 326–336 (1998).
Brändén, M., Sigurdson, H., Namslauer, A., Gennis, R. B., Ädelroth, P. & Brzezinski, P. On the role of the K-proton transfer pathway in cytochrome c oxidase. Proc Natl Acad Sci USA 98, 5013–5018 (2001).
Faxén, K. & Brzezinski, P. The inside pH determines rates of electron and proton transfer in vesicle-reconstituted cytochrome c oxidase. Biochim Biophys Acta 1767, 381–386 (2007).
Namslauer, I., Lee, H. J., Gennis, R. B. & Brzezinski, P. A pathogenic mutation in cytochrome c oxidase results in impaired proton pumping while retaining O2-reduction activity. Biochimica et Biophysica Acta–Bioenergetics 1797, 550–556 (2010).
DeLano, W. L. The PyMOL Molecular Graphics System DeLano Scientific, Palo Alto, CA, USA (2002).
Acknowledgements
PB would like to thank Shelagh Ferguson-Miller, Michigan State University, for valuable discussions. The study was supported by grants from the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Author information
Authors and Affiliations
Contributions
J.V. and A.-L.J. performed experiments. J.V. and P.B. wrote the main manuscript text and prepared figures. P.B. planned research. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vilhjálmsdóttir, J., Johansson, AL. & Brzezinski, P. Structural Changes and Proton Transfer in Cytochrome c Oxidase. Sci Rep 5, 12047 (2015). https://doi.org/10.1038/srep12047
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep12047
This article is cited by
-
Proton-transfer pathways in the mitochondrial S. cerevisiae cytochrome c oxidase
Scientific Reports (2019)
-
Characterization of the quinol-dependent nitric oxide reductase from the pathogen Neisseria meningitidis, an electrogenic enzyme
Scientific Reports (2018)
-
Rapid Electron Transfer within the III-IV Supercomplex in Corynebacterium glutamicum
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
