
Abstract
Coronaviruses (CoVs) continuously threaten human health. However, to date, the evolutionary mechanisms that govern CoV strain persistence in human populations have not been fully understood. In this study, we characterized the evolution of the major antigen-spike (S) gene in the most prevalent human coronavirus (HCoV) OC43 using phylogenetic and phylodynamic analysis. Among the five known HCoV-OC43 genotypes (A to E), higher substitution rates and dN/dS values as well as more positive selection sites were detected in the S gene of genotype D, corresponding to the most dominant HCoV epidemic in recent years. Further analysis showed that the majority of substitutions were located in the S1 subunit. Among them, seven positive selection sites were chronologically traced in the temporal evolution routes of genotype D and six were located around the critical sugar binding region in the N-terminal domain (NTD) of S protein, an important sugar binding domain of CoV. These findings suggest that the genetic drift of the S gene may play an important role in genotype persistence in human populations, providing insights into the mechanisms of HCoV-OC43 adaptive evolution.
Similar content being viewed by others

Introduction
Coronaviruses (CoVs) are widely found in humans as well as in mammalian and avian species, causing asymptomatic infections or respiratory tract disorders and gastroenteritis of varying severity1. CoVs belong to the genus Coronavirus of family Coronaviridae and are classified into four genera, Alphacoronavirus (α-CoV), Betacoronavirus (ß-CoV), Gammacoronavirus and Deltacoronavirus based on phylogenetics and serology1. To date, α-CoV [including human CoV (HCoV)-229E and NL63] and ß-CoV [including HCoV-OC43, HKU1, Severe Acute Respiratory Syndrome CoV (SARS-CoV) and Middle East Respiratory Syndrome CoV (MERS-CoV)] are known to infect human beings1.
Interspecies transmission is a common phenomenon for CoVs that may be responsible for generation of new CoV epidemics during viral evolution2,3,4,5. For instance, the feline CoV (FCoV) type II, a member of α-CoV, might be generated by a double recombination between FCoV type I and canine CoV4. Interestingly, porcine hemagglutinating encephalomyelitis virus, bovine CoV (BCoV) and HCoV-OC43 of ß-CoV are thought to have evolved from the same common ancestor5. During evolution, high frequencies of homologous RNA recombination and gene mutations are considered the main forces that push CoVs to adapt to specific hosts. Such events can lead to emergence of new strains or genotypes within a certain species and even to new species, causing epidemic or zoonotic outbreaks that continuously threaten human health2,3. This phenomenon is exemplified by the recent emergence of SARS-CoV and MERS-CoV6,7. However, the detailed evolutionary mechanism of interspecies transmission and the persistence of CoVs in specific hosts have yet to be fully elucidated.
CoVs have a positive-sense, single-stranded RNA genome, with a length of ~27–31 Kbs1. The spike (S) protein of CoVs protruding on the surface of virions is the major antigenic protein for inducing neutralizing antibodies. However, it is in turn under the highest selection pressure among the viral proteins1,8. S proteins are often cleaved into S1 and S2 subunits to achieve receptor binding and membrane fusion, respectively1. The S1 subunit is composed of two distinct domains, the N-terminal domain (NTD) and the C-terminal domain (CTD), which play important roles in receptor binding1. The NTD is responsible for sugar receptor binding in some CoVs, such as BCoV and HCoV-OC43, or for protein receptor binding in murine hepatitis virus1,9,10,11. The CTD functions as the protein receptor binding domains (RBD) for most of the CoVs1. The S1 subunits of all CoV genera have similar topological structure, preserved sugar-binding functions, but different receptors-binding functions, which suggest that subtly adaptive mutations occur in functional domains during evolution of CoVs12,13. Similar adaptive amino acids mutations around the receptor-binding region have also been found in norovirus, contributing to its epidemic in humans14. Investigation on the evolutionary insights of the S gene, particularly the functional domains, is imperative for understanding the evolution of CoVs and for tracing spillover events and ecological niches.
HCoV-OC43 is the most prevalent CoV in humans and the relatively abundant number of clinical cases and corresponding epidemiological data make it a good model for HCoV adaption evolution1,5,9,15,16,17. Although five genotypes (A to E) have been identified, genotype D has been the dominant OC43 genotype from 2004 to 201215,17. Previous studies by our group and others have demonstrated that recombination contributes to the generation of new OC43 genotypes15,17, but little is known about how HCoV-OC43 genotypes persist in human populations. It is assumed that the continuous adaption of viral antigenic gene is required for the persistence of OC43 genotypes18. However, this hypothesis has not been carefully examined by precise evolutionary pattern analysis. In the present study, we characterized HCoV-OC43 evolution based on the phylodynamic and phylogenetic analysis of full-length S genes to provide insights into its transmission and the adaption.
Results
Relative effective population sizes of OC43 genotypes
To verify the epidemic history of OC43 genotypes over the study period, the relative effective viral population size over time was inferred by analyzing the genetic diversity of the S gene using the Bayesian skyline model. Only the strains obtained from clinical samples containing the full length S, RdRp and N genes retrieved from PubMed (http://www.ncbi.nlm.nih.gov) and GenBank (http://www.ncbi.nlm.nih.gov) for the years 2003 to 2012 were used (see Supplementary Table S1 on line). A varying population size profile was observed (Fig. 1). The overall plot of OC43 (including genotypes B, C, D and E) showed two large bottlenecks. The first was found between 2006 and 2007, showing a decrease in relative genetic diversity in 2006, followed by an increase in 2007. The second was found between 2008 and 2009, with a transient decrease before 2008, followed by an increase in relative genetic diversity coinciding with a temporal epidemic in population size. Genotype B exhibited a transient increase in 2004, then followed a steady but slowly increased relative genetic diversity, corresponding to global increases in its detection17. Genotype D showed a decreased genetic diversity before 2007, then an increase after 2009, consistent with its dominant epidemic from 2007 to 201217. Genotype C and E both exhibited a short and steady relative genetic diversity during the study period. The history of the relative genetic diversity obtained here corresponds to global epidemic data, indicating that the relative genetic diversity measured by the S gene generally reflects the HCoV-OC43 dynamic in population size. The relative genetic diversity of genotype D corresponded to the two bottlenecks of OC43, indicating that genotype D accounts for much of the genetic diversity of OC43 and is the predominant genotype.

Relative genetic diversity dynamics of OC43 and each genotype.
Population size was determined using sequences of 96 OC43 S genes obtained from 2003 to 2012. The median estimates (g) are represented by the black lines and 95% high posterior densities are shown in the color regions.
Evolutionary rate of OC43 genotypes
To explore why genotype D became dominant after 2007, we calculated the evolutionary rate of OC43 genotypes. Using the constant population size under a relaxed-clock method, the mean evolutionary rate of the S gene was estimated to be 8.48 × 10−4 substitutions/site/year for OC43, consistent with previous reports8. For genotypes B, C, D and E, the mean evolutionary rate of S gene was 9.85, 4.85, 8.83 and 6.01 × 10−4 substitutions/site/year, respectively (Table 1). Similar results were obtained using the exponential growth model (Table 1). The highest evolutionary rates were observed in genotype B and D, suggesting that the two genotypes evolved faster than others.
Natural selection of the S gene
To determine whether positive selection took place during the evolution of OC43, the dN/dS values and positive selection amino acids (aa) were calculated. The mean dN/dS ratio was observed in genotype D (0.31), followed by genotypes B (0.29), C (0.20) and E (0.15) (Table 2). Calculations for positive selection sites with a probability (Pr) of > 0.5 in the S gene identified 25 residues in genotype D, six sites in genotype B, one in genotype C, but none in genotype E (Table 2). Seven positive selection sites with Pr of > 0.9 were identified in genotype D. Further analysis showed that 12 of 16 positive selection sites of genotype D and 3 of 5 genotype B in S1 were located at the NTD (aa 15–312, reference strain 5240/07 KF572844), while four positive selection sites in genotype D and one site in genotype B were found in the predicted RBD (aa 339-549, reference strain 5240/07 KF572844)15.
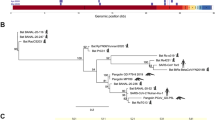
The relatively long epidemic time and more available sequences of genotype D allowed us to analyze in detail the nonsynonymous changes throughout the evolutionary history of OC43. The ancestral sequences were reconstructed and the occurrence time of each nonsynonymous substitution was estimated in the temporal evolution routes using the maximum clade credibility (MCC) tree. The residues at positions 33, 90, 93, 120, 184, 195 and 521 were positive selection sites, with six (33, 90, 93, 120, 184 and 195) located in NTD and one (521) in the predicted RBD. All but one strain (5445/07) identified after 2007 contained the mutation of Y521H. All the strains identified after 2008 contained the mutation K90L. The mutations L195S and Y521H were found in strains identified between 2008 and 2011, but not in those of 2012. Other mutations, including N33D, T93K, D120H and K184N, were found in several strains. The chronologically traced positive sites might be associated with the ladder-like MCC phylogeny of the virus, indicating the key role of aa substitutions in the population dynamics of the virus (Fig. 2).

Molecular clock analysis of OC43 genotype D.
The complete S gene sequences sampled from 2007 to 2012 were used to reconstruct the phylogeny. The calculated positive selection sites in each node are drawn on the MCC tree.
To confirm whether aa substitutions exist in NTD of other genotypes over time, the aa sequences of S from the 12 genotype B and 17 genotype C strains were also aligned. Eleven aa site mutations were observed in genotype B relating to the strain’s isolated over time and seven sites were located in NTD (see Supplementary Fig. S1 online). No aa mutations were found in genotype C over time.
Discussion
Our analysis on the global epidemic of OC43 in recent years showed the temporal transition of genotypes17. It is striking that the evolution of genotype D of OC43 is epochal among the four epidemic genotypes. First identified in 2004, it was generated by recombination15; after a stasis period in 2005 and 2006, a dominant epidemic was found over a longer timescale. As no new recombinant events were detected in the subsequently identified genotype D strains17, the mutations of the S gene−the major antigenic gene, likely plays an important role in driving viral epidemics18.
It is interesting that the positive selection sites calculated in S gene from different isolates over time are corresponding to the genetic variability of genotypes in this study. Genotypes B and D, which have high genetic variability contained more selections sites than that of C and E genotypes. The possible link between the positive selection sites and the genetic variability of genotypes should be evaluated in the future studies. The evaluation of relative genetic diversity, substitution rate, dN/dS value and positive selection sites further showed that genotype D had a significant influence on the relative genetic diversity of OC43 and that its S gene evolved towards heterogeneity. More positive selection sites were also found in the S1 subunit of genotype D. Amino acid substitutions in the surface proteins are considered an important adaption strategy for the persistence of a virus to evade host immune pressure8,18. Although the neutralizing epitopes have not been identified, the substitutions identified in the S1 subunit of OC43 in this study are predicted to be in the antigenicity region (data not shown), which may allow viruses to escape host neutralizing antibodies19,20,21,22. Collectively, these findings suggest that genetic drift may play an important role in maintaining the spread of genotype D in the human population. Whether the genetic variations affect the related antigenic phenotype will need to be confirmed by antigen analysis using S genes with such mutants.
Most of the positive selection sites in S1 subunit are mapped in NTD. Molecular clock tree of genotype D also showed that 6/7 of the predicted substitution were located in NTD. The aa substitutions in NTD seem to be a common evolutionary strategy for CoVs, as the high variability in NTD has also been observed in BCoV and HCoV-NL6321,22. However, this observation warrants further investigation.
BCoV shares a high nucleotide and antigenic similarity with OC4323. The conserved sugar-binding sites identified in BCoV_NTD were also conserved in that of OC43 (see Supplementary Fig. S2 online), indicating the conservation of the core motif in NTD during the species-cross transmission and evolution in human hosts. It is interesting to point out that the evolutionary dynamic pattern of the conserved core and variable outer-region in NTD is similar to that of RBD observed in ß-CoV, suggesting that these functional domains retain some of the ancient records during viral evolution12.
The sugar moieties near the CoV receptor are considered critical co-factors to CoV infection. Antigenic analysis has predicted that there are some epitopes in this domain. Whether the aa mutations around the functional region of NTD are relevant to subtle remodeling of the binding process or antigenic evolution, like that observed in norovirus and influenza virus need to be investigated further20,24,25.
The RBD of OC43 has been predicted15. We found that the positive selection sites in RBD are less than those present in NTD. Notably, a Y521H substitution was found in the genotype D strains identified from 2008 to 2011. The significance of this mutation is unclear. It has been reported that a single aa substitution in RBD can cause marked antigenic differences and enable the virus to escape host immunity in influenza virus and norovirus24,25. Whether this single aa mutation can influence the host receptor binding activity needs to be investigated further. It is interesting that after 2012, the genotype D strains contained less mutations than those identified before 2012. The impact of these changes on the viral prevalence needs continuous surveillance of the OC43 genotypes in the future.
In summary, we report a model for the persistence of OC43 genotypes in human populations based on the first intensive evolutionary analysis of the S gene. We infer that the genetic drift of the S gene is likely to be one of the mechanisms of the adaptation evolution of HCoV-OC43. These findings provide insights into the evolution of CoVs and may have implications in the surveillance of HCoV infections.
Methods
Sequences and phylogenetic analysis
The full length sequences of OC43 S gene available in GenBank (http://www.ncbi.nlm.nih.gov) were retrieved on 30 May 2013 and analyzed together with the sequences identified previously by our group17. A total of 96 full-length S gene sequences obtained from clinical samples were used for analysis. The sequences were aligned using Clustal W program implement in MEGA 5.126. The genotypes of these sequences were determined as reported15,17, including 12 genotype B, 18 genotype C, five genotype E and 61 genotype D. The background information of the sequences including accession numbers, collection dates, isolation areas and genotypes can be found as Supplementary Table S1 online.
Evolutionary analysis
The demographic histories and evolution rates of different OC43 genotypes were determined based on S gene sequence data by the Bayesian Markov Chain Monte Carlo (Bayesian MCMC) method implemented in BEAST (v1.8.1), using a relaxed molecular clock (uncorrelated lognormal-distributed model)27. The best substitution models were selected using Modeltest (version3.7) according to Akaike information criterion (AIC)28. The constant size and exponential growth tree models were used for the inference. Each Bayesian MCMC analysis was run for 100 million states and sampled every 2,000 states. Posterior probabilities were calculated using Tracer (version 1.5). The trees were annotated by the Tree Annotator program implemented in the BEAST package and the MCC tree was visualized using Figtree software (version 1.3.1). Bayesian skyline plots for OC43 genotypes were estimated to depict the relative viral genetic diversity over time.
Positive selection analysis
To infer the positive selection sites of S gene at aa level, the deduced aa sequence entropy was determined using BioEdit (version 7.2.5)29. The ratios of nonsynonymous (dN) to synonymous (dS) substitution were estimated to evaluate the selection pressures on the OC43 S genes, using the codon-based phylogenetic method in CODEML (distributed in PAML, version 4)30. Posterior probabilities of the inferred positive selection sites were calculated using the Bayes empirical Bayes (BEB) approach which accounts for sampling errors31. The chronological evolution of dN changes throughout the evolutionary history of OC43 genotype D was traced using HyPhy software (version 2.2) and dN substitution was estimated using the maximum clade credibility (MCC) tree generated from Bayesian MCMC molecular clock analysis27,32. The aa sites were positioned according to the HCoV-OC43_D 5240/07 (KF572844).
Additional Information
How to cite this article: Ren, L. et al. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci. Rep. 5, 11451; doi: 10.1038/srep11451 (2015).
References
Masters, P. S. & Perlman, S. Coronaviridae. Fields Virology. 6thEd. Knipe, D. M. & Howley, P. M. (ed.) 825–854. Lippincott Williams & Wilkins, Philadelphia, 2013.
Coleman, C. M. & Frieman, M. B. Coronaviruses: important emerging human pathogens. J Virol 88, 5209–5212 (2014).
Woo, P. C., Huang, Y., Lau, S. K. & Yuen, K. Y. Coronavirus genomics and bioinformatics analysis. Viruses 2, 1804–1820 (2010).
Motokawa, K., Hohdatsu, T., Hashimoto, H. & Koyama, H. Comparison of the amino acid sequenceand phylogenetic analysis of the peplomer, integral membrane and nucleocapsid proteins of feline, canine and porcine coronaviruses. Microbiol Immunol 40, 425–433 (1996).
Vijgen, L. et al. Evolutionary history of the closely related group 2 coronaviruses: porcine hemagglutinating encephalomyelitis virus, bovine coronavirus and human coronavirus OC43. J Virol 80, 7270–7274 (2006).
Drosten, C. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348, 1967–1976 (2003).
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367, 1814–1820 (2012).
Bush, R. M. Predicting adaptive evolution. Nat Rev Gene 2, 387–392 (2001).
Schultze, B., Gross, H. J., Brossmer, R. & Herrler, G. The S protein of bovine coronavirus is a hemagglutinin recognizing 9-O-acetylated sialic acid as a receptor determinant. J Virol 65, 6232–6237 (1991).
Künkel, F., & Herrler, G. Structural and functional analysis of the surface protein of human coronavirus OC43.Virology 195, 195–202 (1993).
Schultze, B. et al. Transmissible gastroenteritis coronavirus, but not the related porcine respiratory coronavirus, has a sialic acid (N-glycolylneuraminic acid) binding activity. J Virol 70, 5634–5637 (1996).
Li, F. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J Virol 86, 2856–2858 (2012).
Lu, G. et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500, 227–231 (2013).
Bok, K., et al. Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J Virol 83, 11890–901 (2009).
Lau, S. K. et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J Virol 85, 11325–11337 (2011).
Vijgen, L. et al. Circulation of genetically distinct contemporary human coronavirus OC43 strains. Virology 337, 85–92 (2005).
Zhang, Y. et al. Genotype shift in human coronavirus OC43 and emergence of a novel genotype by natural recombination. J Infect 70, 641–650 (2015)
Pybus, O. G. & Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat Rev Genet 10, 540–550 (2009).
Chibo, D. & Birch, C. Analysis of human coronavirus 229E spike and nucleoprotein genes demonstrates genetic drift between chronologically distinct strains. J Gen Virol 87, 1203–1208 (2006).
Dominguez, S. R. et al. Genomic analysis of 16 Colorado human NL63 coronaviruses identifies a new genotype, high sequence diversity in the N-terminal domain of the spike gene and evidence of recombination. J Gen Virol 93, 2387–2398 (2012).
Bidokhti, M. R. et al. Evolutionary dynamics of bovine coronaviruses: natural selection pattern of the spike gene implies adaptive evolution of the strains. J Gen Virol 94, 2036–2349 (2013).
Cavanagh, D., Davis, P. J. & Mockett, A. P. Amino acids within hypervariable region 1 of avian coronavirus IBV (Massachusetts serotype) spike glycoproteinare associated with neutralization epitopes. Virus Res 11, 141–150 (1988).
Peng, G. et al. Crystal structure of bovine coronavirus spike protein lectin domain. J Biol Chem 287, 41931–41938 (2012).
Donaldson, E. F., Lindesmith, L. C., Lobue, A. D. & Baric, R. S. Norovirus pathogenesis: mechanisms of persistence and immune evasion in humanpopulations. Immunol Rev 225, 190–211 (2008).
Lewis, N. S. et al. Substitutions near the hemagglutinin receptor-binding site determine the antigenic evolution of influenza A H3N2 viruses in U.S. swine. J Virol 88, 4752–4763 (2014).
Tamura, K. et al. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Molecular Biology and Evolution 28, 2731–2739 (2011).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7, 214 (2007).
Posada, D. & Crandall, K. A. MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818 (1998).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41, 95–98 (1999).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol BiolEvol 24, 1586–1591 (2007).
Yang, Z. et al. Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22, 1107–1118 (2005).
Pond, S. L., Frost, S. D. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679 (2005).
Acknowledgements
We would like to thank Dr. Patrick CY Woo and Dr. Susanna KP Lau (The University of Hong Kong, Hong Kong SAR, China) for providing sequence and primer information. This study was supported in part by the National Major Science & Technology Project for Control and Prevention of Major Infectious Diseases in China (2012ZX10004-206, 2014ZX10004-001), the Program for New Century Excellent Talents in University (3332013127), the China National Funds for Distinguished Young Scientists (81225014), Program for Changjiang Scholars and Innovative Research Team in University (IRT13007) and Fondation Mérieux.
Author information
Authors and Affiliations
Contributions
L.L.R., Y.Z., G.P.B. and J.W.W. conceived and designed experiments. L.L.R., Y.Z., J.G.L., Y. X., J. Z., Y. W. and L. C. performed the experiments. L.L.R., Y.Z., J.G.L., Y. X. and J.W.W. analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ren, L., Zhang, Y., Li, J. et al. Genetic drift of human coronavirus OC43 spike gene during adaptive evolution. Sci Rep 5, 11451 (2015). https://doi.org/10.1038/srep11451
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11451
This article is cited by
-
Epitope similarity cannot explain the pre-formed T cell immunity towards structural SARS-CoV-2 proteins
Scientific Reports (2020)
-
Identification and evolutionary dynamics of two novel human coronavirus OC43 genotypes associated with acute respiratory infections: phylogenetic, spatiotemporal and transmission network analyses
Emerging Microbes & Infections (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
