
Abstract
Direct optimization of the metabolic pathways on the chromosome requires tools that can fine tune the overexpression of a desired gene or optimize the combination of multiple genes. Although plasmid-dependent overexpression has been used for this task, fundamental issues concerning its genetic stability and operational repeatability have not been addressed. Here, we describe a rapid and reliable strategy for chromosomal integration of gene(s) with multiple copies (CIGMC), which uses the flippase from the yeast 2-μm plasmid. Using green fluorescence protein as a model, we verified that the fluorescent intensity was in accordance with the integration copy number of the target gene. When a narrow-host-range replicon, R6K, was used in the integrative plasmid, the maximum integrated copy number of Escherichia coli reached 15. Applying the CIGMC method to optimize the overexpression of single or multiple genes in amino acid biosynthesis, we successfully improved the product yield and stability of the production. As a flexible strategy, CIGMC can be used in various microorganisms other than E. coli.
Similar content being viewed by others

Introduction
Significant progress has been achieved in the area of metabolic engineering and synthetic biology with the development of genetic methods that allow the manipulation of homogenous and heterologous DNA in microorganisms. Meanwhile, optimizing the overexpression of rate-limiting genes is a necessary step to direct more carbon flux into the biosynthesis pathway and to prevent the accumulation of toxic intermediate metabolites. To accomplish this task, plasmid-based overexpression has been extensively applied because of its easy manipulation and regulated expression1,2,3. However, this strategy suffers from genetic instability resulting from segregational instability, structural instability and allele segregation4. Moreover, to maintain the existence of plasmids in host cells, antibiotic or other selective agents should be utilized, which increases the overall bioprocess cost and generates environmental concerns5.
Integration of the target genes into the host chromosome is a preferable strategy to overcome the drawbacks of plasmid-based overexpression. In Escherichia coli, homologous recombination, site-specific recombination and transposon-mediated gene transposition are often used to achieve chromosomal integration. In the case of homologous recombination, a single crossover between a targeting gene and a homologous DNA fragment on a chromosome can be realized with the aid of the endogenous RecA and RecBCD complex6,7,8. When the three genes gam, bet and exo originating from λ phage, were employed, the targeting gene bordered with short-length (35–50 bp) homologous arms was more effectively integrated into the chromosome than the RecA-based recombination9,10,11,12. However, the recombination frequency dropped sharply when the targeting DNA exceeded 2.5 kb in size13,14. Thus, Sabri et al.15 constructed a series of knock-in/knock-out vectors to integrate large targeting DNA sequences into the chromosome. Employing this strategy, a 7.3-kb DNA fragment at one locus and an 11.3-kb DNA fragment across two loci were integrated.
Transposon, which is a mobile genetic element, was also previously used to integrate heterologous DNA into the bacterial chromosome16,17. On the basis of transposon, Wei et al.18 successively integrated three resistance genes into the chromosome of E. coli BW25113. The location sites of the transposon insertion were evenly distributed on the chromosome. To identify these location sites, genomic DNA digestion with NheI, self-ligation with T4 ligase and DNA sequencing were required. Apart from homologous recombination and transposon-mediated gene transposition, site-specific recombination provides another effective solution to introduce a target DNA fragment into a specific chromosome site19. Haldimann and Wanner20 achieved efficient integration by constructing a series of conditional-replication, integration and modular plasmids that contained the phage integration genes int and xis. However, the entire integration process requires 1 to 2 weeks to complete because of the numerous recombination steps. To reduce the operation time and steps, St-Pierre et al.21 recently developed a novel method named “clonetegration” to integrate DNA into prokaryotic chromosomes.
Although several chromosomal integration strategies have been extensively reported, little attention has been paid to the integration of gene(s) with multiple copies. The only typical example in the literature is a chemically inducible chromosomal evolution (CIChE) system22. Through multiple rounds of evolution with increased antibiotic concentrations, an engineered strain with 40–60 copies of target genes on the chromosome was obtained via recA-dependent homologous recombination. Recently, a flippase (FLP)-dependent recombination from the yeast 2-μm plasmid has attracted our attention23. The only requirements for FLP-dependent recombination are a FLP recombinase and a 34-bp FLP recombination target (FRT) site. The FLP/FRT recombination system has mainly been used to excise selective markers in the Red recombination system24. However, because FLP recombinase catalyzes a reversible reaction, it could also be used to integrate DNA into the chromosome. Thus, in the present study, we developed a strategy for the chromosomal integration of gene(s) with multiple copies (CIGMC) based on FLP recombinase. With this method, multiple copies of a single gene or multiple genes can be stably integrated into the chromosome in one step.
Results
Development of the CIGMC system based on FLP/FRT recombination
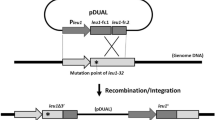
The CIGMC system was developed based on FLP recombinase, which can catalyze recombination between an FRT-containing circular DNA and an FRT-containing linear DNA (or genome DNA). Thus, the targeting strain in this strategy should possess at least one FRT site on the chromosome, which can be easily introduced by the Tn5 transposon25. In this study, GPT101, a previously constructed L-tryptophan-producing strain containing four FRT sites on the chromosome, was selected26. The recA gene, which encodes the DNA strand exchange and recombination protein, was then deleted in GPT101 to prevent possible homologous recombination. This generated an additional FRT site. Therefore, the final targeting strain, named GPF-5, contains five FRT sites on the chromosome. The construction outline of CIGMC was presented in Fig. 1.

Outline of the chromosomal integration of gene(s) with multiple copies (CIGMC) system in Escherichia coli.
recA, encoding the DNA strand exchange and recombination protein, was previously deleted to prevent subsequent homologous recombination that could reduce the integrated copy number. kan, kanamycin resistance gene; R6K, narrow-host-range replicon; gfp, green fluorescent protein; FRT, flippase recombination target.
The integrative plasmid pG-1 was constructed by ligating the FRT site, gfp and kan in vitro. To avoid the replication of this plasmid in the target strain during recombination, which may result in false-positive clones, an integrative vector without a replicon was initially used. The constructed integrative plasmid pG-1 was then electroporated into the host strain GPF-5 and the integrants were visually screened on plates and then fluorescently analyzed in microtiter plates. The relative fluorescence units (RFU) per OD600 of the integrants spanned from 114.3 to 495.0 (Fig. 2a). This observation demonstrated that pG-1 was successfully integrated into the chromosome at various copy numbers. Using quantitative polymerase chain reaction (qPCR), we proved that the integrated copy number ranged from 1–8 and with an average of 2.36 (Fig. 2b). Of the 150 detected strains, 63 had only one copy of pG-1 integrated and only two strains had eight copies integrated (Fig. 2c). This possibly due to the relatively low final concentration (2.5 ng/μL) of the integrative plasmid pG-1.

Characterization of the CIGMC system by randomly integrating pG-1 into the chromosome of strain GPF-5.
(a) Relative fluorescence units (RFU) per OD600 of the 150 CIGMC strains randomly selected. (b) Integrated copy number of pG-2 of the CIGMC strains in Fig. 2a. NA indicates the average integrated copy number. (c) Distribution of the integrated copy number of the 150 CIGMC strains in Fig. 2a. Error bars represent the s.d. (n = 3).
To improve the final concentration of the integrative plasmid, a narrow-host-range replicon, R6K, was inserted into plasmid pG-1, generating plasmid pG-2. Because replicon R6K is strictly dependent on the π protein encoded by the pir gene, the integrative plasmid pG-2 can only replicate in strains carrying pir, for instance, E. coli BW2514127,28. Thus, a high concentration of pG-2 can be isolated for integration from E. coli BW25141 containing pG-2. Using pG-2, the maximal fluorescence intensity of the integrants increased from 495.0 to 675.9 RFU/OD600, which is equal to a copy number of 12 (Fig. 3). The average copy number also increased from 2.4 to 3.75, suggesting a positive correlation between the integrative plasmid concentration and the copy number of the integrated gene(s) on the chromosome. To obtain a strain containing a high copy number of target genes, a high concentration of integrative plasmid appears to be a prerequisite.

Characterization of the CIGMC system by randomly integrating pG-2 into the chromosome of strain GPF-5.
(a) RFU per OD600 of the 150 CIGMC strains randomly selected. (b) Integrated copy number of pG-2 of the CIGMC strains in Fig. 3a. NA indicates the average integrated copy number. (c) Distribution of the integrated copy number of the 150 CIGMC strains in Fig. 3a. Error bars represent the s.d. (n = 3).
Factors that affect the integrated copy number of CIGMC
The relationship between the integrated copy number and the final concentration of integrative plasmid was analyzed (Table 1) and a positive correlation between them was identified. The maximum integrated copy number reached 15 and the average integrated copy number was 5.41 when 40 ng/μL of pG-2 was applied. Further increases in plasmid concentration did not improve the integrated copy number, indicating that this reaches saturation. For general operation, 30 ng/μL of integrative plasmid is sufficient for CIGMC. Additionally, the increased number of FRT sites on the chromosome was predicted to improve CIGMC efficiency because of the increased availability of integration sites. To demonstrate this, we selected five strains with 1–5 FRT sites respectively as CIGMC targets. As shown in Table 2, an average integrated copy number of 2.28 and a maximum integrated copy number of five were obtained for the strain with only one FRT site. Increasing the number of FRT sites from two to five on the chromosome in single increments resulted in a maximum integrated copy number that increased from 5 to 7, 9, 10 and 12, while the average integrated copy number increased from 2.28 to 2.59, 2.81, 3.29 and 3.75, respectively. This result demonstrated that increasing the number of FRT sites on the chromosome is beneficial to CIGMC and that there is an approximately linear correlation between the number of FRT sites and the average integrated copy number.
Application of CIGMC to optimize gene overexpression
Because CIGMC generated a library of integrants with various copy numbers of target gene(s), this method would be ideal for optimizing the overexpression of the key gene in a metabolic pathway. A tryptophan-producing strain, GPT1002, was constructed previously by our group26. Three genes, tktA, aroGFR and trpEFR, were overexpressed in this strain. Another enzyme shikimate kinase, encoded by aroK, was proven to have a pivotal function in aromatic amino acid biosynthesis29. An optimized overexpression of aroK was supposed to improve the L-tryptophan production. However, plasmid-based overexpression of aroK did not obtain a positive result. Therefore, we attempted to optimize the overexpression of aroK with CIGMC. The integrated copy number of aroK was similar to that using GFP as a CIGMC target (Fig. 4a and Supplementary Fig. S1). All CIGMC strains exhibited similar growth status with the control, indicated by the maximum OD600 achieved after 24 h batch cultivation. Single-copy integration of aroK on the chromosome improved L-tryptophan production per OD600 from 0.159 to 0.214 g/L. Two integrated copies of aroK further improved the L-tryptophan production to 0.298 g/L per OD600, which was 87.4% higher than that of the initial strain. However, L-tryptophan production sharply dropped when the integrated copy number further increased. This demonstrates that the presence of two integrated copies of aroK is optimal and explains why plasmid-based overexpression was unsuccessful. Together, these findings suggest that CIGMC provides an optimization strategy to directly regulate gene overexpression on the chromosome, especially when single-copy integration with strong promoter and ribosome binding site (RBS) is not sufficient.

Application of the CIGMC strategy to optimize amino acid production in E. coli.
(a) L-tryptophan accumulation of CIGMC strains by introducing random copies of aroK into GPF-5 and transforming with plasmid pTAT. The L-tryptophan production was determined after batch cultivation for 24 h. The indicated copy number excludes the original aroK gene on the chromosome. L-tryptophan-producing strain GT-1, containing wild-type aroK, deleted recA and plasmid pTAT, was selected as a control. (b) Integrated copy number of serAFR in CIGMC strains. (c) Integrated copy number of serB in CIGMC strains. (d) Integrated copy number of serC in CIGMC strains. (e) L-serine production of CIGMC strains with random copies of serAFR, serB and serC integrated into the chromosome. Strain GPF-11, containing deleted recA and plasmid pYF-1 overexpressing serAFR, serB and serC, was selected as a control. The L-serine production was determined after batch cultivation for 36 h. (f) Stability of genetic constructs and L-serine production in recombinant strain Y-7, which has 10 copies of serAFR, 4 copies of serB and 4 copies of serC. Error bars represent the s.d. (n = 3).
Application of CIGMC to optimize the metabolic pathway
In a previous study, we constructed a recombinant E. coli strain that can produce 0.38 g/L L-serine per OD600 in batch cultivation by overexpressing serAFR, serB and serC, encoding deregulated 3-phosphoglycerate dehydrogenase, phosphoserine aminotransferase and phosphoserine phosphatase respectively, in a medium-copy plasmid30. However, the L-serine production was unstable because of plasmid instability. To test if CIGMC could provide an alternative overexpression strategy, serAFR, serB and serC were combined as an artificial operon in recombinant plasmid pG-7 (Supplementary Fig. S2) and were integrated into the chromosome together. Direct integration of this operon into the chromosome by CIGMC generated integrants with a relatively low integrated copy number and the maximum L-serine production was only 0.183 g/L per OD600 (Supplementary Fig. S3). This indicated that either the operon was too large or that the three genes needed to be expressed at different levels. Thus, we designed an independent integration strategy, wherein all three genes were simultaneously integrated with separate integrative plasmids pG-4, pG-5 and pG-6. In this case, serAFR, serB and serC were integrated into the host at different copy numbers. As shown in Fig. 4b–4d, a strain library with a combination of different copies of the three genes was generated in one step and the optimal L-serine-producing strain could then be screened from the library.
The fermentation results showed that strains Y-7, Y-22 and Y-23 exhibited higher L-serine production than the control strain GPF-11, which was constructed by the plasmid-based overexpression of serAFR, serB and serC (Fig. 4e and Supplementary Fig. S4). Strain Y-7 produced 0.425 g/L/OD600 L-serine, which is the highest among the 40 strains detected. qPCR analysis indicated that it contained 10 copies of serAFR, four of serB and four of serC. Another strain, containing two copies of serAFR, two of serB and three of serC, only produced 0.132 g/L/OD600 L-serine. Analysis of all integrants indicated that strains with high copies of serAFR produced more L-serine, suggesting that serAFR is crucial for E. coli L-serine production. Additionally, different CIGMC strains exhibited similar maximum OD600 to each other, indicating that little additional metabolic burden was generated as the copy number of target genes increased on the chromosome (Supplementary Fig. S4). We next tested the stability of the engineered strain and demonstrate that the genetic constructs and L-serine production of Y-7 were stable after 10 rounds of subculture without antibiotics (Fig. 4f). By contrast, plasmid pYF-1 in GPF-11 was completely lost after six rounds of subculture without antibiotics, leading to a sharp decrease in L-serine production (Supplementary Fig. S5).
In this study, we firstly used quantitative reverse transcription PCR (qRT-PCR) to verify the copy number of target genes (Supplementary Fig. S6–S8). However, the instability of RNA may influence the accuracy of the results. Thus, we then chose qPCR instead by directly using genome DNA as templates (Figure 2–4). Although most results were similar, some differences were also existence in the copy numbers obtained by these two methods, indicating qPCR was a better choice to determine the copy number of target gene(s) in CIGMC.
Discussion
In this study, a CIGMC strategy based on FLP/FRT site-specific recombination was developed. Using this strategy, we successfully integrated 15 copies of a single gene or 18 total copies of three genes into the chromosome of E. coli in one step; the only requirements were a target strain with recA deletion and FRT sites on the chromosome.
The initially constructed integrative plasmid did not contain a replicon. Thus, the concentration of donor plasmid used for integration was low. To increase its concentration, we introduced a narrow-host replicon, R6K, which can replicate in strains harboring the π protein. This approach significantly increased the concentration of integrative plasmid and the integration efficiency. Yu et al.31 also reported that the targeting efficiency increased in a near-linear relationship with increasing concentrations of donor DNA from 0–300 ng in the λ recombination system of E. coli. FLP recombinase can catalyze excision, inversion, integration, or translocation in response to different substrates. FLP/FRT recombination consists of four main steps: DNA binding, synapsis, recombination and dissociation32. The in vitro study of Ringrose et al.33 demonstrated that the conversion rate of the FLP-bound substrate DNA into the excised synaptic complex (k34) is 2.45-fold higher than that of the reverse process (k-34). Thus, they suggested that the preferred direction of FLP was excision. However, in our study, a high integrative plasmid concentration was used. When the final concentration of pG-2 was 20 ng/μL, each recipient cell would receive at least 1.34 × 104 copies of the integrative plasmid. [Because there were approximately 5 × 105 E. coli cells in 1 μL of competent cell solution, the relative molecular mass of the integrative plasmid pG-2 was 1.85 × 106 and the Avogadro constant is 6.02 × 1023, each recipient competent cell would receive 20 × 10−9 × 6.02 × 1023/(1.85 × 106 × 5 × 105) = 1.34 × 104 copies of pG-2 when 20 ng of pG-2 was applied]. Even when only partial pG-2 could be electroporated into the competent cells, the in vivo pG-2 concentration should be much higher than that of the chromosomal DNA, thus guaranteeing the occurrence of integration. Increasing the number of FRT sites on the chromosome also improved the integration efficiency and the average copy number (Table 2). This result is similar to the integration at the attB/attP site catalyzed by ΦC31 integrase34. We deduced that the increased number of FRT sites may improve the binding opportunity of FLP recombinase.
FLP/FRT recombination was previously used to integrate exogenous DNA at defined locations in E. coli25,35. However, this attempt was not further investigated, probably because FLP/FRT site-specific recombination has no obvious advantages over other recombination strategies. On the other hand, there is a pressing need to optimize the overexpression of gene(s) directly on the chromosome because of the lack of chromosome integration tools in this field. The common optimization strategies of gene overexpression include constructing a plasmid with different copy numbers or different strengths of promoters36,37,38, ribosome-binding sites39,40, or terminators41. Strains that contain various plasmids or plasmid libraries can then be screened with respect to their phenotypes. Libraries of promoters, ribosome-binding sites, or terminators can also be achieved on the chromosome using novel technologies such as multiplex automated genome engineering (MAGE)42,43. However, the expression level of the target gene cannot exceed the maximum strength of the promoters, which is normally too weak for overexpression. Moreover, the screening of such libraries is both tedious and difficult.
The CIGMC method provides a strategy to directly optimize the overexpression of a single gene or multiple genes in a metabolic pathway on the chromosome with several advantages. First, the direct integration of genes is more stable and reliable compared with plasmid-based overexpression. A previous study demonstrated that a recombinant strain harboring a medium-copy plasmid completely lost the ability to produce polyhydroxybutyrate (PHB) after 35 generations even in the presence of antibiotics, whereas the PHB accumulation of the recombinant strain derived from chromosome integration was stable without antibiotics for a long period22. In addition, as shown in Supplementary Figure S1 and Supplementary Figure S4, different CIGMC strains exhibited similar maximum OD600, indicating little additional metabolic burden was generated as the increased copy number of target genes on the chromosome. Second, a similar effect can be achieved with fewer copies of genes on the chromosome compared with plasmid-based overexpression. In our constructed amino acid producing strains, strain Y-7, with an average integrated copy number of six per gene, could produce more L-serine than the recombinant strain that harbors a plasmid of medium copy number (15–20 copies). In some cases, when plasmids with tandem promoters were employed, it was normally too weak for overexpression44, but the integration of target genes into the chromosome can overcome this problem. Finally, CIGMC can be used to integrate several genes simultaneously and to generate a gene(s) integration library in which every targeted gene has a random copy number on the chromosome. From this library, an optimized overexpression combination of several genes in the metabolic pathway can be obtained after screening. In our experiment, strain Y-7, which had 10 copies of serAFR, four of serB and four of serC, exhibited the best combination of the three genes and produced the most L-serine (0.425 g/L/OD600).
As an effective genome integration method, CIChE also exhibited several advantages for chromosomal integration. The maximum integrated copy number achieved by CIChE was 40–60, which is substantially higher than CIGMC. Moreover, CIChE could realize the integration of large operons with high copies, while only three copies of the serAFR-serB-serC operon were integrated into the chromosome by CIGMC. However, CIGMC can generate a strain library with combinations of different copy number from multiple genes in one step. By using 96-well plates and the high-throughput screening method, integrants with the desired phenotype can be selected in 3–4 days. By contrast, at least several weeks were needed for CIChE due to the chromosomal evolution process. Therefore, CIChE is fit for integrating ≥ 15 copies of target gene(s), while CIGMC is a better choice for the quick integration of 1–15 copies of target genes(s) and exploring the optimal integrated copy number of a single gene or multiple genes.
In summary, CIGMC directly generates a library of integrants with various copies of different genes integrated on the chromosome, which can be used to optimize the overexpression of gene(s). For single-gene integration together with GFP, fluorescence-activated cell sorting would be a good choice. To screen candidate integrants with a combination of multiple genes, online monitored microtiter plates are necessary. Because FRT/FLP recombination can function well in bacteria45 and fungi46, CIGMC is a flexible strategy that can be applied in other microorganisms.
Methods
Bacterial strains
All strains used in this study are listed in Supplementary Table S1. E. coli strains DH5α and BW25141 were used as the hosts of recombinant DNA manipulation.
Plasmid construction
The plasmids and oligonucleotides used in this study are listed in Supplementary Tables S2 and S3, respectively. To construct plasmid pG-1, the FRT-kan-trc-gfp module was constructed using primers p-F/p-R and template plasmid pLYK. This DNA fragment was then digested with XhoI and self-ligated using T4 DNA ligase. To construct integrative plasmid pG-2, the FRT-kan-trc-gfp module and narrow-host-range replicon R6K were amplified using primers KG-F/KG-R and R6K-F/R6K-R and pG-1 and pKD4 were selected as templates. These two fragments were then assembled and cyclized with the one-step sequence- and ligation-independent cloning (SLIC) method47.
To integrate random copies of aroK into the genome of GPF-5, three DNA fragments, R6K-FRT-kan, lac promoter and aroK, were generated using OFK-F/OFK-R, lac-F/lac-R and aroK-F/aroK-R as the primers and plasmid pG-2, pCL1920 and the genome of wild-type E. coli W3110 as the templates, respectively. Next, these three fragments with 30–40 homologous bases were assembled and cyclized by the SLIC method to generate pG-3. Plasmid pG-4 was obtained in the same way by assembling and cyclizing three fragments: R6K-FRT-kan, the lac promoter and serAFR.
To obtain integrative plasmid pG-5, two template plasmids, pCLB and pCLC, were initially constructed. Two genes involved in the L-seine biosynthesis pathway, serB and serC, were amplified by primers serB-F/serB-R and serC-F/serC-R, respectively and the genome of wild-type E. coli W3110 was selected as the template. Individual double-digestion by HindIII and PstI was then performed for serB and serC and pCLB and pCLC were generated by ligating these two DNA fragments into pCL1920, respectively. Afterwards, four DNA fragments, R6K-FRT, trc promoter, aadA1 gene and lac-serB, were amplified by primers OF-F/OF-R, trc-F/trc-R, aadA1-F/aadA1-R and LSB-F/LSB-R and template plasmids pG-1, pLYK, pCL1920 and pCLB, respectively. These four fragments with 30–40 homologous bases were assembled using the SLIC method to obtain pG-5. Plasmid pG-6 was constructed through the same method except that fragments aadA1 and lac-serB were separately replaced by tetA and lac-serC. To construct plasmid pG-7, three DNA fragments, FRT-kan, R6K and trc-serAFR-serB-serC, were amplified using primers G-1F/G-1R, G-2F/G-2R and G-3F/G-3R and template plasmids pG-1, pG-2 and pYF-1, respectively. Then, these three fragments were also assembled and self-cyclized using the SLIC method.
Gene deletion and insertion
To prevent subsequent homologous recombination, which could reduce the copy number of integrated gene(s), recA, encoding a DNA strand exchange and recombination protein with protease and nuclease activity, was deleted in GPT101 using the one-step inactivation method24. Primers recA-F/recA-R and template plasmid pKD4 were used to obtain the linearized DNA flanked by homologous sequences. Electroporation was then conducted according to the manufacturer's instructions. Positive clones on the plates were verified by PCR using the primers recA-TF/recA-TR and the kanamycin cassette was removed by the helper plasmid pCP20 to obtain strain GPF-5. In addition, recA was also deleted in wild-type W3110, GPT98, GPT99, GPT100 and L-serine-producing strain YF-6 through the same method. To verify the integrated copy number of candidate strains, three control strains with only one copy of the relative resistant gene were constructed. Primers kan1-F/kan1-R, spc1-F/spc1-R and tet1-F/tet1-R, as well as template plasmids pKD4, pG-5 and pG-6, were used to obtain the linearized DNA of kan, aadA1 and tetA for recombination, respectively. Next, the three fragments separately replaced the recA gene of YF-6. After electroporation and overnight cultivation, positive clones on the plates were verified by PCR using the primers recA-TF/recA-TR. The control strains GPF-7, GPF-8 and GPF-9 were thus obtained.
Integration of gene(s) by CIGMC
To integrate gene(s) with multiple copies, plasmid pCP20 overexpressing the FLP recombinase was first transformed into the target strain. A single clone of this target strain was pre-cultivated in 5 mL Luria-Bertani medium (1% tryptone, 0.5% yeast extract and 1% NaCl) at 30°C and on a rotary shaker at 250 rpm overnight. Afterwards, 1 mL of the overnight cells were inoculated into 50 mL SOB medium (2% tryptone, 0.5% yeast extract, 0.05% NaCl, 2.5 mM KCl and 10 mM MgCl2) and cultivated at 30°C to an OD600 of 0.5. Strains were then cultivated at 42°C to induce the expression of FLP recombinase for 20–30 min. Integrative plasmid was then electroporated into the host cells and 1 mL SOC medium (2% tryptone, 0.5% yeast extract, 0.05% NaCl, 2.5 mM KCl, 10 mM MgCl2 and 20 mM glucose) was added to shocked cells and incubated for 1 h at 42°C. Strains with the relative antibiotic-resistant phenotype were selected on plates. The integrated copy number was roughly estimated using the fluorescent method and accurately determined by qPCR.
To investigate the influence of the FRT site number on the efficiency of CIGMC, GPF-1, GPF-2, GPF-3, GPF-4 and GPF-5, containing one, two, three, four and five FRT sites on their chromosomes, respectively, were used in the CIGMC reaction. To explore the influence of integrative plasmid concentration on the CIGMC efficiency, integrative plasmid pG-2 was used with different final concentrations ranging from 5 ng/μL to 50 ng/μL. The final concentration of integrative plasmid in CIGMC was defined as the mass of supplemented integrative plasmid per unit volume of competent cells.
Process of strain construction using CIGMC
An L-tryptophan-producing strain was constructed by introducing the integrative plasmid pG-3 containing the FRT-kan-lac-aroK module into the genome of GPF-5. The copy number of aroK was determined using qPCR with GPF-10 containing one copy of kan as a control. L-tryptophan production was then evaluated using the integrants with different copies of aroK and plasmid pTAT containing the L-tryptophan synthesis genes.
An L-serine-producing strain was constructed by introducing integrative plasmid pG-7 containing the serAFR-serB-serC module into the genome of GPF-6. The integrated copy number was determined using qPCR. To realize an independent integration of genes serAFR, serB and serC, the three integrative plasmids pG-4, pG-5 and pG-6 were constructed and simultaneously integrated into the chromosome of GPF-6 by CIGMC. GPF-7, GPF-8 and GPF-9 were selected as control strains to determine the integrated copy number of serAFR, serB and serC, respectively. The L-serine production of the integrants with different combinations of serAFR, serB and serC was then evaluated.
qPCR analysis
The integrated copy number of target gene(s) was detected by qPCR on genomic DNA isolated from the candidate strains using the TIANamp Bacteria DNA Kit (Tiangen Biotech, Beijing, China). qPCR was performed with SYBR Premix Ex TaqII (Takara Bio, Dalian, China) following the protocol of the LightCycler 480 RT-PCR System (Roche, Basel, Switzerland). Primers used in qPCR are listed in Supplementary Table S3.
Growth conditions
Strains for cloning and inoculation were grown in Luria-Bertani medium at 37°C for 8–12 h. Ampicillin (100 mg/L), chloramphenicol (17 mg/L), kanamycin (25 mg/L), spectinomycin (50 mg/L), or tetracycline (20 mg/L) was supplemented when necessary. Isopropyl β-D-1-thiogalactopyranoside was supplemented at the final concentration of 0.2 mM. For L-tryptophan production, the seed medium contained (per liter): 20 g glucose, 5 g MgSO4·7H2O, 1.5 g KH2PO4, 10 g (NH4)2SO4, 15 g yeast extract, 15 mg FeSO4·7H2O, 0.5 g sodium citrate dehydrate and 100 mg vitamin B1. The fermentative medium contained (per liter): 20 g glucose, 5 g MgSO4·7H2O, 2 g KH2PO4, 4 g (NH4)2SO4, 1 g yeast extract, 100 mg FeSO4·7H2O and 2 g sodium citrate dehydrate. For batch fermentation of L-serine, the mineral AM1 medium48 supplemented with 1 g/L yeast extract and 20 g/L glucose was used. For preliminary fermentation screening, 96-well microtiter plates were incubated in a deep-well bioshaker (Bio-Xplorer, Beijing, China). In addition, the genetic stability was evaluated as described previously22.
Analytical methods
Cell growth was monitored at OD600 with a spectrophotometer (Shimazu, Kyoto, Japan). L-tryptophan was determined using the fluorometric method49. L-serine was quantitatively analyzed using high-performance liquid chromatography (Shimadzu) equipped with a Venusil AA column (250 mm × 4.6 mm, Agela Technologies, USA). The fluorescence of integrants was determined in 96-well microtiter plates as described previously44.
References
Juminaga, D. et al. Modular engineering of L-tyrosine production in Escherichia coli. Appl Environ Microbiol 78, 89–98 (2012).
Jiang, M. & Pfeifer, B. A. Metabolic and pathway engineering to influence native and altered erythromycin production through E. coli. Metab Eng 19, 42–49 (2013).
Alper, H., Fischer, C., Nevoigt, E. & Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A 102, 12678–12683 (2005).
Friehs, K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol 86, 47–82 (2004).
Hägg, P., De Pohl, J. W., Abdulkarim, F. & Isaksson, L. A. A host/plasmid system that is not dependent on antibiotics and antibiotic resistance genes for stable plasmid maintenance in Escherichia coli. J Biotechnol 111, 17–30 (2004).
Court, D. L., Sawitzke, J. A. & Thomason, L. C. Genetic engineering using homologous recombination. Annu Rev Genet 36, 361–388 (2002).
Posfai, G., Koob, M. D., Kirkpatrick, H. A. & Blattner, F. R. Versatile insertion plasmids for targeted genome manipulations in bacteria: isolation, deletion and rescue of the pathogenicity island LEE of the Escherichia coli O157:H7 genome. J Bacteriol 179, 4426–4428 (1997).
Martinez-Morales, F., Borges, A. C., Martinez, A., Shanmugam, K. T. & Ingram, L. O. Chromosomal integration of heterologous DNA in Escherichia coli with precise removal of markers and replicons used during construction. J Bacteriol 181, 7143–7148 (1999).
Song, C. W. & Lee, S. Y. Rapid one-step inactivation of single or multiple genes in Escherichia coli. Biotechnol J 8, 776–784 (2013).
Baumgartner, F., Seitz, L., Sprenger, G. A. & Albermann, C. Construction of Escherichia coli strains with chromosomally integrated expression cassettes for the synthesis of 2'-fucosyllactose. Microb Cell Fact 12, 40 (2013).
Yang, J. et al. High-efficiency scarless genetic modification in Escherichia coli using Lambda-Red recombination and I-SceI cleavage. Appl Environ Microbiol 80, 3826–3834 (2014).
Song, J., Dong, H., Ma, C., Zhao, B. & Shang, G. Construction and functional characterization of an integrative form lambda Red recombineering Escherichia coli strain. FEMS Microbiol Lett 309, 178–183 (2010).
Ublinskaya, A. A., Samsonov, V. V., Mashko, S. V. & Stoynova, N. V. A PCR-free cloning method for the targeted ϕ80 Int-mediated integration of any long DNA fragment, bracketed with meganuclease recognition sites, into the Escherichia coli chromosome. J Microbiol Methods 89, 167–173 (2012).
Kuhlman, T. E. & Cox, E. C. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res 38, e92 (2010).
Sabri, S., Steen, J. A., Bongers, M., Nielsen, L. K. & Vickers, C. E. Knock-in/Knock-out (KIKO) vectors for rapid integration of large DNA sequences, including whole metabolic pathways, onto the Escherichia coli chromosome at well-characterised loci. Microb Cell Fact 12, 60 (2013).
Haniford, D. B. & Chaconas, G. Mechanistic aspects of DNA transposition. Curr Opin Genet Dev 2, 698–704 (1992).
Mizuuchi, K. Transpositional recombination: mechanistic insights from studies of Mu and other elements. Annu Rev Biochem 61, 1011–1051 (1992).
Wei, X. X. et al. A mini-Mu transposon-based method for multiple DNA fragment integration into bacterial genomes. Appl Microbiol Biotechnol 87, 1533–1541 (2010).
Sauer, B. Site-specific recombination: developments and applications. Curr Opin Biotechnol 5, 521–527 (1994).
Haldimann, A. & Wanner, B. L. Conditional-replication, integration, excision and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183, 6384–6393 (2001).
St-Pierre, F. et al. One-step cloning and chromosomal integration of DNA. ACS Synth Biol 2, 537–541 (2013).
Tyo, K. E., Ajikumar, P. K. & Stephanopoulos, G. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat Biotechnol 27, 760–765 (2009).
Senecoff, J. F., Bruckner, R. C. & Cox, M. M. The FLP recombinase of the yeast 2-micron plasmid: characterization of its recombination site. Proc Natl Acad Sci U S A 82, 7270–7274 (1985).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645 (2000).
Huang, L.-C., Wood, E. A. & Cox, M. M. Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J Bacteriol 179, 6076–6083 (1997).
Gu, P., Yang, F., Kang, J., Wang, Q. & Qi, Q. One-step of tryptophan attenuator inactivation and promoter swapping to improve the production of L-tryptophan in Escherichia coli. Microb Cell Fact 11, 30 (2012).
Skrzypek, E., Haddix, P. L., Plano, G. V. & Straley, S. C. New suicide vector for gene replacement in yersiniae and other gram-negative bacteria. Plasmid 29, 160–163 (1993).
Taylor, R. K., Manoil, C. & Mekalanos, J. J. Broad-host-range vectors for delivery of TnphoA: use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J Bacteriol 171, 1870–1878 (1989).
Ikeda, M. Towards bacterial strains overproducing L-tryptophan and other aromatics by metabolic engineering. Appl Microbiol Biotechnol 69, 615–626 (2006).
Gu, P. et al. Construction of an L-serine producing Escherichia coli via metabolic engineering. J Ind Microbiol 41, 1443–1450 (2014).
Yu, D. et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97, 5978–5983 (2000).
Stark, W. M., Boocock, M. R. & Sherratt, D. J. Catalysis by site-specific recombinases. Trends Genet 8, 432–439 (1992).
Ringrose, L. et al. Comparative kinetic analysis of FLP and cre recombinases: mathematical models for DNA binding and recombination. J Mol Biol 284, 363–384 (1998).
Xie, F. et al. Adjusting the attB site in donor plasmid improves the efficiency of ΦC31 integrase system. DNA Cell Biol 31, 1335–1340 (2012).
Huang, L. C., Wood, E. A. & Cox, M. M. A bacterial model system for chromosomal targeting. Nucleic Acids Res 19, 443–448 (1991).
Jensen, P. R. & Hammer, K. Artificial promoters for metabolic optimization. Biotechnol Bioeng 58, 191–195 (1998).
Jorgensen, C. M., Hammer, K., Jensen, P. R. & Martinussen, J. Expression of the pyrG gene determines the pool sizes of CTP and dCTP in Lactococcus lactis. Eur J Biochem 271, 2438–2445 (2004).
Khlebnikov, A., Risa, Ø., Skaug, T., Carrier, T. A. & Keasling, J. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J Bacteriol 182, 7029–7034 (2000).
Levin-Karp, A. et al. Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. ACS Synth Biol 2, 327–336 (2013).
Chen, S. et al. Automated design of genetic toggle switches with predetermined bistability. ACS Synth Biol 1, 284–290 (2012).
Chen, Y. J. et al. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods 10, 659–664 (2013).
Wang, H. H. et al. Genome-scale promoter engineering by coselection MAGE. Nat methods 9, 591–593 (2012).
Wang, H. H. et al. Programming cells by multiplex genome engineering and accelerated evolution. Nature 460, 894–898 (2009).
Li, M. et al. A strategy of gene overexpression based on tandem repetitive promoters in Escherichia coli. Microb Cell Fact 11, 19 (2012).
Ishikawa, M. & Hori, K. A new simple method for introducing an unmarked mutation into a large gene of non-competent gram-negative bacteria by FLP/FRT recombination. BMC Microbiol 13, 86 (2013).
Bloemendal, S. et al. Tools for advanced and targeted genetic manipulation of the beta-lactam antibiotic producer Acremonium chrysogenum. J Biotechnol 169, 51–62 (2014).
Jeong, J. Y. et al. One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl Environ Microbiol 78, 5440–5443 (2012).
Martinez, A. et al. Low salt medium for lactate and ethanol production by recombinant Escherichia coli B. . Biotechnol Lett 29, 397–404 (2007).
Iizuka, H. & Yajima, T. Fluorometric determination of L-tryptophan with methoxyacetaldehyde. Biol Pharm Bull 16, 103–106 (1993).
Acknowledgements
This research was financially supported by grants from the National Natural Science Foundation of China (31370085), the National High-Tech Research and Development Plan of China (2012AA022104), China Postdoctoral Science Foundation funded project (2014M551894) and Special Funds of Shandong Postdoctoral Innovation Project (201402026).
Author information
Authors and Affiliations
Contributions
P.G. carried out most of the experiments. F.Y. constructed the L-serine producing strains and performed qPCR. F.Y. and T.S. implemented batch fermentation. Q.Q., P.G., Q.W. and Q.L. drafted the manuscript. Q.Q. conceived and supervised the study. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gu, P., Yang, F., Su, T. et al. A rapid and reliable strategy for chromosomal integration of gene(s) with multiple copies. Sci Rep 5, 9684 (2015). https://doi.org/10.1038/srep09684
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep09684
This article is cited by
-
Metabolic engineering of Escherichia coli for the production of (R)-α-lipoic acid
Biotechnology Letters (2023)
-
Toward improved terpenoids biosynthesis: strategies to enhance the capabilities of cell factories
Bioresources and Bioprocessing (2022)
-
Advances and prospects in metabolic engineering of Escherichia coli for L-tryptophan production
World Journal of Microbiology and Biotechnology (2022)
-
Genomic landscapes of bacterial transposons and their applications in strain improvement
Applied Microbiology and Biotechnology (2022)
-
Parallel multi-objective optimization approaches for protein encoding
The Journal of Supercomputing (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
