
Abstract
In primary brain tumors, oncogenes are frequently amplified and maintained on extrachromosomal DNA as double minutes (DM), but the underlying mechanisms remain poorly understood. We have generated a mouse model of malignant glioma based on knock-in of a mutant PDGF receptor α (PDGFRα) that is expressed in oligodendrocyte precursor cells (OPCs) after activation by a Cre recombinase. In the tumor suppressor INK4/Arf−/− background, mutant animals frequently developed brain tumors resembling anaplastic human gliomas (WHO grade III). Besides brain tumors, most animals also developed aggressive fibrosarcomas, likely triggered by Cre activation of mutant PDGFRα in fibroblastic cell lineages. Importantly, in the brain tumors and cell lines derived from brain tumor tissues, we identified a high prevalence of DM Pdgfra gene amplification, suggesting its occurrence as an early mutational event contributing to the malignant transformation of OPCs. Amplicons extended beyond the Pdgfra locus and included in some cases neighboring genes Kit and Kdr. Our genetically defined mouse brain tumor model therefore supports OPC as a cell of origin for malignant glioma and offers an example of a defined temporal sequence of mutational events, thus providing an entry point for a mechanistic understanding of DM gene amplification and its functionality in gliomagenesis.
Similar content being viewed by others


Introduction
Focal amplification of genomic DNA is a pathological hallmark in many solid tumors, including glioblastoma, the most frequent and deadly form of primary brain cancer1. Amplicons typically comprise 0.5–10 MB of DNA and exist either as circular extrachromosomal structures termed double minutes (DM), or as intrachromosomal concatenated repeats termed homogenously staining regions (HSR)2. These two forms of gene amplification are interrelated and possibly interconvertable3. Several models have been proposed on how DM arise during chromosome replication, including replication fork stalling, fork collision and fragmentation of larger chromosomal segments3,4,5,6. However, mechanistic details of DM formation and its functionality in cancer development remain poorly understood. For instance, despite high prevalence in glioblastoma2, it is unclear whether DM gene amplification occurs as an early mutational event contributing to malignant transformation, or at a later stage in advanced glioblastomas as a result of global genomic instability7,8.
Increased activity of the receptor tyrosine kinase (RTK) PDGF receptor α (PDGFRα) is frequently encountered in glioblastoma and commonly associated with amplification of the PDGFRA gene9,10,11,12,13,14. Additionally, PDGFRA amplification is often associated with point mutations or structural variants that are thought to increase the intensity of PDGFRα signaling (Refs. 14, 15; http://www.cbioportal.org). A number of mouse glioma models have been generated by overexpressing secreted PDGF ligands in the brain (for review, see Refs. 16, 17), but since secreted PDGF ligands may act through both autocrine and paracrine mechanisms, the cell of origin cannot be conclusively defined in these glioma models. These models also do not address the prevalence of PDGFRA gene amplification and its oncogenic role in gliomagenesis. Providing an adequate animal model to investigate these fundamental questions is central to advancing glioma research and therapeutic options.
We have developed a new glioma mouse model based on cell-autonomous activation of PDGFRα in oligodendrocyte precursor cells (OPCs). Specifically, we utilized a conditional knock-in of a PDGFRα point mutation that reduces auto-inhibition of the kinase domain18. The knock-in design ensures that the expression of mutant PDGFRα is under the control of the endogenous Pdgfra promoter, which, in the CNS, is active in OPCs19. When bred on INK4/Arf−/− background, a common tumor suppressor deletion in glioblastoma, mice developed spontaneous primary brain tumors between 15–30 weeks of age. Brain tumors displayed pathological characteristics of human high grade gliomas (WHO grade III), thus providing direct evidence for OPC as a cell of origin for malignant glioma. Importantly, we identified a high prevalence of amplification of the Pdgfra mutant allele as DM in not only advanced, but also early stage grade III gliomas, thus supporting RTK amplification as an important early event in the malignant transformation of OPCs. In summary, our study describes a novel glioma model that provides an example of a defined temporal sequence of mutational events in the malignant transformation of OPCs, starting from an activating RTK mutation and loss of a tumor suppressor, followed by amplification of the mutant RTK in the form of DM.
Results
A glioma mouse model driven by PDGFRα overactivity
To generate a glioma mouse model based on cell-autonomous activation of PDGFRα in OPCs, we utilized mice that carry conditional knock-in alleles of PDGFRα with the point mutations V561D (termed J for “juxtamembrane”) or D842V (termed K for “kinase domain”)18,20 (Fig. 1a). Both point mutations confer kinase overactivity due to reduced autoinhibition, with the K mutation being to some extent a stronger activator than the J mutation18. The PDGFRα J or K mutant alleles are expressed from the endogenous Pdgfra promoter after Cre recombinase-mediated excision of a STOP cassette. We crossed the conditional mutant mice to Nestin-Cre or GFAP-Cre transgenic lines, leading to Cre activation of mutant PDGFRα in all forebrain areas (Fig. S1). The knock-in design restricts in the CNS PDGFRα J/K expression to OPCs, which endogenously express PDGFRα19,21. Animals with overactive PDGFRα did not exhibit higher numbers of OPCs or mature oligodendrocytes (Fig. S2a). Furthermore, OPCs isolated from mutants proliferated at a normal rate in vitro (Fig. S2b) and mutant cohorts developed no signs of brain tumors, indicating that PDGFRα J/K mutation by itself is insufficient to increase OPC proliferation or drive gliomagenesis.

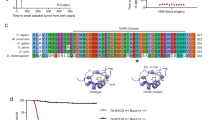
PDGFRα driven brain tumor model.
(a) Schematic diagram of PDGFRα J/K knock-in alleles. ATG, start codon; SA, splice acceptor; STOP, PGK-neo cassette. (b) Kaplan-Meier survival curves of 4 mouse mutant cohorts with brain tumors. Mice generally succumbed to subcutaneous fibrosarcomas and brain tumors were detected by histological analysis. (c) Example of early stage tumor growth, as revealed by IHC for proliferation marker Ki67 and PDGFRα. Note high density of Ki67+ proliferating cells in tumor area, increased expression level of PDGFRα and invasive migration of tumor cells through corpus callosum into contralateral hemisphere. (d) H&E staining of an advanced brain tumor growth (asterisk in tumor centre, dashed line demarcates expansion). Scale bars: 50 μm (c, d).
To facilitate the incidence of brain tumors, we bred PDGFRα J/K mutant mice onto the background of INK4A/Arf−/−, a frequent tumor suppressor mutation in glioblastoma1. Mice bearing both PDGFRα J/K and INK4A/Arf−/− mutations developed normally until early adulthood and displayed normal numbers of OPC (Fig. S3a, b). Interestingly, at ages of 15–30 weeks, an estimated 10% of the animals in the GFAP-Cre; PdgfrαK/+; INK4A/Arf−/− cohort displayed an enlarged skull and apparent neurological symptoms (Fig. S3c), indicative of advanced primary brain tumors (see below). It is noteworthy that about 90% of mice in this GFAP-Cre activated cohort and 100% in the Nestin-Cre; PdgfrαK/+ or J/+; INK4A/Arf−/− cohort were affected by aggressive subcutaneous fibrosarcomas (Fig. S3c), presumably derived from Nestin-Cre or GFAP-Cre activation of PDGFRα J/K in fibroblastic cell lineages in the skin (Fig. S1a). The histological characteristics of the fibrosarcomas, including overexpression of Pdgfra and high grade features such as pleomorphic nuclei and numerous mitotic figures, were virtually identical to tumors that were previously described in detail in a PdgfrαK/+; INK4A/Arf−/− tumor model with general Cre activation18. Consequently, most cohort mice in our study had to be sacrificed due to fast growing fibrosarcomas before brain tumors could reach larger sizes and our subsequent studies were mostly conducted on gliomas at relatively early stages of brain tumor growth.
Sacrificed mice were screened for signs of neoplastic growth in brains by immunostaining for PDGFRα and proliferation marker Ki67 (see Fig. 1b for survival curves and brain tumor incidence). More than 50% of animals in the mutant cohorts GFAP-Cre; PdgfraK/+; INK4A/Arf−/− and Nestin-Cre; PdgfraJ/+; INK4A/Arf−/− harbored highly proliferative areas in the brain, reflecting early (Fig. 1c and S4a) and occasionally more advanced stages of brain tumor growth (Fig. 1d; Fig. S4b). In the Nestin-Cre; PdgfraK/+; INK4A/Arf−/− cohort, mice generally succumbed to fibrosarcomas by 15 weeks, before brain tumors were detectable (Fig. S3a). In the background of heterozygous INK4A/Arf deletion (Nestin-Cre; PdgfraK/+; INK4A/Arf+/−), animals developed brain tumors only rarely and cohort mice with heterozygous INK4A/Arf mutation alone (INK4A/Arf+/−) never developed brain tumors (Fig. S3a). Various regions of the forebrain were affected by tumor growth, including cortex, striatum and thalamus (Fig. 1c, d; Fig. S4). All brain tumors displayed a highly invasive phenotype, with preferential migration routes along fiber tracts in the corpus callosum (Fig. 1c). We also observed a consistent molecular feature of increased immunointensity for PDGFRα in brain tumor cells as compared to normal OPCs (Fig. 1c; Fig. S4b), suggesting that not only the kinase overactivity from the J/K point mutation, but also upregulation of PDGFRα expression are required for the malignant transformation of OPCs.
PDGFRα-driven murine brain tumors resemble human grade III gliomas
We analyzed brain tumors histologically by H&E staining, which revealed densely cellular and diffusely infiltrating glial neoplasms, with several histopathological features of anaplastic gliomas (WHO grade III): brisk mitotic activity, hypercellularity, cellular pleomorphism and marked nuclear atypia, i.e. hyperchromatic nuclei with irregular contours (Fig. 2a, b). Tumor cells were often arranged in secondary structures of Scherer, i.e. perineuronal satellitosis, perivascular cuffing, intrafascicular growth and subarachnoid spreading (Fig. 2c–f). At the time of sacrifice, most brain tumors had not reached a size at which necrotic areas became widespread; however, we detected areas of decreased cellularity, rarefaction of neuropil, presence of pyknotic nuclei and apoptotic debris, likely representing incipient necrosis (Fig. 2g). Based on cellularity and morphological appearances, virtually all tumors were comparable with human high grade gliomas (WHO grade III), such as anaplastic oligodendrogliomas or anaplastic astrocytomas, but lacked pseudopalisading necrosis or glomeruloid microvascular proliferation that are diagnostic of WHO grade IV glioblastoma.

PDGFRα driven brain tumors display features of high grade glioma.
(a–g) Histopathological analysis of tumor areas by H&E staining shows a high concentration of mitotic figures (a, arrows), high cellularity and nuclear atypia (b), perineuronal satellitosis (c; N, neuronal nuclei), perivascular growth (d), intrafascicular growth (e), subarachnoid spreading (f) and areas of incipient necrosis (g; arrows point to pyknotic nuclei). (h–k) IF labeling of brain tumor sections for cell type specific markers. Nuclei labeled with DAPI are shown in blue. Tumor cells with high PDGFRα expression were highly proliferative, as seen by proliferation marker Ki67 (h) and express the OPC cell lineage markers Olig2, Sox2, Sox10 and Ng2, as well as the neural stem cell marker Nestin (i–k). Tumor cells were negative for immunosignal of astroglial marker GFAP, mature oligodendrocyte marker APC-CC1 and neuronal marker NeuN (l–n). Scale bars: 10 μm (a–g), 20 μm (h–n).
Further analysis by immunofluorescence revealed several consistent molecular features, such as increased PDGFRα expression, high proliferative index by Ki67 staining (Fig. 2h) and expression of OPC markers Olig2, Sox10, Ng2 and Sox2 (Fig. 2i–k). Additionally, tumor cells express Nestin, a neural stem cell (NSC) marker that is activated during malignant transformation of OPC22 (Fig. 2k). In contrast, markers of differentiated neural cells, such as GFAP (for astrocytes), APC-CC1 (for mature oligodendrocytes) and NeuN (for neurons), were not detectable in brain tumor cells (Fig. 2l–n).
Amplification of PDGFRα K allele in glioma cells
For further analysis on a cellular level, we derived cell lines from forebrain tissue of mutant and control mice and cultured them in neural stem cell media under spherogenic conditions, which allows for selection of the so-called glioma stem cells (GSC; also known as tumor-propagating cells). Cell lines derived from advanced brain tumors are expected to consist predominantly of GSCs, whereas cell lines from wild-type brain will only consist of normal neural progenitors. Cell lines derived from tissue of early stage gliomas with lower numbers of tumor cells may consist of either GSCs, normal neural progenitors, or both cell types.
We established cell lines from mice in our brain tumor cohorts, including 5 mice with advanced grade III gliomas that had resulted in a bulging skull and behavioral impairment and 10 mice with early stage grade III gliomas detected only by histological analysis. To investigate the mechanism of the upregulation of PDGFRα expression in glioma cells, we screened these cell lines for Pdgfra gene copy numbers by qPCR. We observed Pdgfra amplification in 9 of the 15 lines derived from tumor-bearing mice, including all 5 lines from mice bearing advanced gliomas and 4 lines from mice bearing early stage gliomas (Fig. 3a). The average copy number of the Pdgfra gene in these 9 glioma cell lines varied from 4 to 21 copies (Fig. 3a). The other 6 lines from mice bearing early stage gliomas showed no Pdgfra gene amplification. It is worth noting that these 6 lines may consist predominantly of normal neural progenitors and not glioma cells; another possibility is that in these glioma cases alternative mechanisms, such as epigenetic modifications, transcription factor activation, or increased protein stability, might be responsible for the upregulation of PDGFRα expression. As controls, we also screened spherogenic cell lines derived from wild-type mice, INK4A/Arf−/− mice and brain tumor cohort mice with no detectable brain tumors and observed no Pdgfra amplifications (Fig. 3a). Taken together, these data demonstrate that Pdgfra amplification is a frequent event that can occur during early stages of malignant glioma development and may serve as an important mechanism for the upregulation of PDGFRα expression in gliomas.

Gene amplification of PDGFRα in glioma cell lines.
(a) Increased Pdgfra allele copy numbers detected by qPCR in 9 cell lines derived from glioma-bearing brains, but not in control cell lines. PCR primers were either generic for all Pdgfra alleles or specific for Pdgfra J/K mutant alleles. Asterisks denote cell lines in which both mutant and wild-type allele amplifications have occurred. (b) Amplification can also include the adjacent genes Kit (co-amplified in 5 out of 9 lines) and Kdr (in 1 out of 9 lines). Arrowheads or # signs denote co-amplification of Kit or Kdr.
Preferential amplification of the mutant allele of PDGFRα
To distinguish gene amplification of PDGFRα J/K mutant vs. wild-type allele, we utilized qPCR primers specific for the mutant alleles. In the majority of glioma cell lines tested (7 out of 9), the copy numbers of the mutant allele approached the total number of all Pdgfra alleles (note that mutant specific primers can not detect the wild-type Pdgfra allele), indicating a preferential amplification of the PDGFRα J/K mutant allele (Fig. 3a). In the remaining two glioma lines (line #28 and #49), qPCR results showed that both mutant and wild-type alleles were amplified (Fig. 3a).
To determine if the amplification is focally restricted to the Pdgfra gene, or encompasses a larger chromosomal area, we first probed for amplicons 50 kb upstream and downstream of the Pdgfra locus. Of the 6 cell lines tested, 5 lines showed amplification extending beyond the Pdgfra locus by at least 50 kb in both directions and one line showed amplification only downstream of the Pdgfra locus, indicating that amplicons are in general larger than 100 kb (Fig. S5a). Next, we investigated copy numbers of two adjacent RTK genes, Kit and Kdr, located approximately 0.4 Mb and 0.8 Mb downstream of Pdgfra, respectively. Of note, Kit and Kdr are frequently co-amplified with Pdgfra in human glioblastoma23. We found that among the 9 Pdgfra-amplified glioma cell lines tested, Kit was co-amplified in 5 lines and one of these lines also had additional Kdr co-amplification (Fig. 3b). Finally, we analyzed amplification of Mdm2 and Egfr, two of the most commonly amplified oncogenes in glioblastoma1, but located on different chromosomes than Pdgfra. We found only two copies of each gene in all cell lines tested (Fig. S5b), suggesting absence of widespread gene amplification or global genomic instability in this tumor model.
Amplified PDGFRα is maintained as extrachromosomal DM
To determine the cytogenetic nature of the Pdgfra amplification, we conducted fluorescent in situ hybridization (FISH) on metaphase spreads obtained from glioma cell lines using probes for the Pdgfra locus (located on chromosome 5) and for the centromere of chromosome 5 (internal control). In all Pdgfra-amplified glioma cell lines, we found that in addition to the two Pdgfra loci on the chromosome 5 pair, FISH signals were also abundant on small extrachromosomal pieces of DNA (Fig. 4a), corresponding to DM, a class of circular extrachromosomal DNA fragments. We did not detect Pdgfra amplicons integrated into chromosomes. The majority of DM that were visualized by DAPI counterstaining were positive for the Pdgfra FISH signal, but a few DM without Pdgfra amplicons were also detected (Fig. S6).

Double minute amplification of PDGFRα.
(a) Metaphase spreads of 9 Pdgfra amplified cell lines and one wild-type control line, hybridized with FISH probe for PDGFRα locus (red) and chromosome 5 centromere region (green), revealing amplification of PDGFRα locus as extrachromosomal DM. The cell line number is indicated in each image. (b) FISH hybridization on brain tumor sections confirms amplification of PDGFRα locus in situ. Examples of three brain tumors and one unaffected adjacent area are shown. The number of the corresponding cell line is indicated in each image. Chromosomes and nuclei labeled with DAPI are shown in blue. Scale bars: 5 μm (a), 20 μm (b).
The Pdgfra copy numbers in individual cells from the same glioma cell line were highly variable (Fig. S7), likely reflecting unequal DM distribution among daughter cells during cell division24. It also highlights intratumoral heterogeneity of Pdgfra copy number status. We also screened directly sections of brain tumor tissues for Pdgfra amplification by FISH and confirmed widespread presence of multiple Pdgfra amplicons in tumor areas (Fig. 4b), thus supporting the notion that Pdgfra amplification is a driving factor during gliomagenesis.
Discussion
High grade gliomas are thought to be a heterogeneous form of cancer, originating from different neural cell types, including astrocytes, neural stem cells and OPCs25,26. In our study, by activating a mutant PDGFRα cell-autonomously in the OPC population, we provide direct evidence for OPC as a cell of origin for malignant glioma. A series of previous studies have utilized overexpression of PDGF-A or -B in the mouse brain to induce gliomagenesis (reviewed in Refs. 16, 17). Of particular interest are studies that directed PDGF-B overexpression specifically to late OPCs/early oligodendrocytes, which strongly support OPC as cell of origin of glioma27,28. However, due to potential paracrine actions of secreted PDGF ligands on different cell types, the cell of origin could not be unambiguously determined. In contrast, our glioma model is based on expression of a mutant PDGFRα receptor under the control of the endogenous Pdgfra promoter in the OPC population19,21,29. Of note, expression of PDGFRα has also been reported in a population of neurovascular cells21,30, but the molecular features of gliomas observed in our study make it unlikely that this cell type contributed to tumorigenesis. Expression of PDGFRα in neural stem cells in the SVZ, as has been proposed earlier, was not detected in the present study, in agreement with more recent observations29,31.
The expression of mutant PDGFRα J/K in OPC by itself did not lead to gliomagenesis, but when combined with loss of INK4A/Arf, was able to drive gliomagenesis. This result is reminiscent of studies with mutant EGF receptor, which by itself triggers gliomagenesis only at very low levels and requires additional deletion of INK4A/Arf for frequent glioma formation32,33. These studies therefore suggest that loss-of-function mutations of INK4A/Arf or other tumor suppressor genes may be an early permissive event that facilitates transforming gene amplifications. A significant finding of our study is the spontaneous DM amplification of Pdgfra, preferentially of the mutant allele, in early stage gliomas. Hence, our model provides an example where two preexisting mutations–activating RTK mutation and INK4A/Arf deletion–facilitate tumorigenesis through a third mutational event, that is, amplification of the mutant RTK.
In human primary glioblastomas, which arise de novo, PDGFRA amplifications are found in 10–20% of cases, which makes PDGFRA the second most frequently amplified RTK gene in glioblastoma after EGFR34,35. Among primary glioblastomas of the proneural non-CpG island methylation (non G-CIMP) subtype, PDGFRA amplifications even reach a frequency of greater than 50%1. On the other hand, Secondary glioblastomas, which transform gradually from low grade G-CIMP gliomas, are characterized by expression of a proneural profile with high PDGF pathway activity36. Further studies will determine how well our glioma model of Pdgfra DM amplification is suited to study development of primary proneural glioblastomas, secondary glioblastomas, or both.
The challenge of studying gene amplification using patient-derived samples is that they invariably harbor numerous genetic mutations and display vast intratumoral heterogeneity, making it difficult to determine the conditions permissive for gene amplification. Intratumoral heterogeneity of gene amplification is further reflected by the fact that many glioblastomas carry DM amplifications of either PDGFRA or EGFR in intermingled tumor cell populations23,34,35. Our mouse glioma model provides a unique entry point for studying the propensity of glioblastoma to amplify RTKs, as our model is genetically defined with only two initiating mutations–a point mutant of PDGFRα with elevated kinase activity and loss of INK4A/Arf.
The high prevalence of Pdgfra amplification in our model suggests that Pdgfra amplification can occur as an early spontaneous event. This is in contrast to earlier concepts that gene amplification takes place in advanced tumors after accumulation of a large number of mutations that ultimately facilitate genomic amplifications7. In further support, PDGFRA amplification is present in more than 15% of human grade III anaplastic astrocytoma and anaplastic oligodendroglioma, consistent with PDGFRA amplification as an early transforming event during glioma progression34. The fact that we observed preferential amplification of the mutant but not the wild-type allele of Pdgfra implies that the kinase activity of the amplified PDGFRα is under positive selection during gliomagenesis. If Pdgfra amplification were simply a result of genomic instability, both the wild-type and the mutant alleles of Pdgfra would be amplified at similar frequencies.
Our approach of cultivating tumor cell lines allowed preparations of metaphase spreads that provided direct evidence for Pdgfra amplification as DM. We also confirmed that amplicons extend beyond the Pdgfra locus, resembling the situation in human glioblastoma23. Interestingly, in a mouse glioma model generated by loss of two tumor suppressors, PTEN and p53, spontaneous focal amplifications of RTK genes EGFR, MET, or PDGFRα as DM were described37. This study and ours demonstrate that loss of tumor suppressors such as Ink4A/Arf, or PTEN and p53, create a condition in which additional focal amplifications of RTKs can drive malignant transformation of glioma cells.
DM gene amplifications are numerically unstable in proliferating cell populations due to variable replication and uneven segregation between daughter cells. In our PDGFRα-driven glioma model, the copy numbers of PDGFRα vary among individual tumor cells. In the case of EGFR amplification in glioblastoma, mathematical modeling suggests that low level EGFR amplification is an early event, whereas high level amplification is a late event8. Recent modeling also suggests that a potential pathway to glioma encompasses initial PDGF-A overexpression followed by PDGFRA amplification at later stages38. Gene copy numbers in tumor cells are perhaps dictated by the need for an optimal tumor growth equilibrium, as excess gene copies may over-intensify signaling flux with detrimental consequences. For instance, tumor cells can dynamically reduce copy numbers of EGFR-vIII DM in response to tyrosine kinase inhibitor (TKI) treatment, but return back to previous levels upon TKI withdrawal39. Future studies will determine whether glioma cells under treatment with PDGFRα specific TKI exhibit a similar adaptive regulation of PDGFRα DM copies. Gene amplification by DM can also be associated with structural rearrangements of oncogenes40, hence further characterization of PDGFRα rearrangements will be needed to elucidate this mechanism. Obtaining a better mechanistic understanding of DM gene amplification and its oncogenic roles in the initiation and progression of glioma may lead to new therapeutic strategies for the treatment of brain tumors.
Methods
Mouse mutant alleles
PDGFRα J/K: Pdgfratm12Sor and Pdgfratm13Sor 18; Nestin-Cre: Tg(Nes-cre)1Kln41; GFAP-Cre: Tg(GFAP-cre)25Mes42; INK4A/Arf knockout: Cdkn2atm1Rdp 43; R26R-lacZ reporter: Gt(ROSA)26Sortm1Sor 44. Brain tumor cohorts were bred on a mixed genetic background of FVB, C57BL/6J and 124S4. Mice were euthanized in accordance with NIH Guidelines for the Care and Use of Laboratory Animals and animal protocols were approved by the IACUC committee of Icahn School of Medicine at Mount Sinai.
Histology
Antibodies and their dilutions used for immunostaining of brain sections: goat anti-PDGFRα (R&D Systems AF1062, 1:50); rabbit anti-PDGFRα (Santa Cruz Biotechnology SC-338, 1:50); rabbit anti-Ki67 (Abcam 15580, 1:500); rabbit anti-Olig2 (Millipore AB9610, 1:500); rabbit anti-Sox2 (Millipore AB5603, 1:200); goat anti-Sox10 (R&D Systems AF2864, 1:50); mouse anti-Nestin (Millipore MAB353, 1:250); rabbit anti-Ng2 (Millipore AB520, 1:500); rabbit anti-NeuN (Novus NBP1-77686SS, 1:200); rabbit anti-GFAP (Invitrogen 180063, 1:200); mouse anti-APC, clone CC1 (Calbiochem OP80, 1:20).
Secondary antibodies were coupled with Alexa fluorophores (Jackson Immunoresearch) or biotinylated for ABC signal amplification and peroxidase DAB staining (Vector Labs).
For mRNA in situ hybridization, cryosections were hybridized with Digoxigenin-labeled riboprobes, which were synthesised from the plasmids pBS-Pdgfra-ISH-1.6 kb and pCR4-Plp1-ISH-2.0 kb and probe hybridization was detected with AP-conjugated anti-Dig Fab fragments (Roche) and NBT/BCIP staining.
Hematoxylin & Eosin staining was performed with Shandon Gill #2 Hematoxylin and Eosin-Y (Thermo Scientific).
Glioma cell lines
The anterior third of the forebrain of mice was dissected and tissue was minced into small pieces with a scalpel blade and then incubated in Accutase (Invitrogen) for 15 min at 37°C. Cells were gently dissociated with a 5 ml pipette and the cell suspension was filtered through a 40 μm cell strainer (BD Falcon) and then cultured in Neural Stemcell media (Stemcell Technologies) for several days until spheres of >200 μm had formed45. Spheres were passaged by Accutase dissociation and archived in liquid nitrogen storage in Neural Stemcell media containing 10% DMSO.
Oligodendroctyte precursor cell culture
Oligodendrocyte precursor cells (OPC) were isolated from cortices of P5 mice by Papain digest and selective immunopanning with anti-PDGFRα antibodies and then cultured in poly-D-lysine coated tissue culture dishes with defined OPC media containing PDGF-A46.
Fluorescence in situ hybridization (FISH)
Cell lines were cultivated over night in presence of 0.1 μg/ml colcemid (Invitrogen) before cytogenetic preparation by KCl treatment and fixation. The mouse BACs RPCI-23-222P8 and RPCI-23-413L22 were used as templates for synthesis of FISH probes for the chromosome 5 centromere and Pdgfra gene locus, respectively (Empire Genomics, Buffalo, NY). Cells and tissue sections were hybridized with FISH probes according to manufacturer's instructions (Empire Genomics) and counterstained with DAPI.
Quantitative PCR
Gene copy numbers were determined by qPCR of genomic DNA from cell lines with a SYBR green reaction mix (Quanta Biosciences) on an ABI7900 device and normalized to a PCR for the Foxp2 locus. Primer sequences (5′-3′): Pdgfra: GGGGAGAGTGAAGTGAGCTG and CATCCGTCTGAGTGTGGTTG; PdgfraJ/K mutant allele only: CAAACTCTTCGCGGTCTTTC and CGAAGTTATATTAAGGGTTCCG; Pdgfra 50 kb upstream: GATGCTGAGACACTCCTCTTG and GACTTGCTTAGTGCATCTGTATTG; Pdgfra 50 kb downstream: GATAGAGGCCCATGTCTTCTTAC and GCAGACTAACATGACAGGAGAA; Egfr: CCTGGAAGAGACCTGCATTATC and GTTAAACCCACTACTGAGACAGG; Kit: CAGCACATAGCCCAGGTAAA and CAACTCTTGCCGAGCTGATA; Kdr: CTCCATGACATAAGGCCTACAC and CTGCCACAAGCCTACTGATAA; Mdm2: GGAAGTCGATGGTTGGGAATAG and AGCTGACAGAGAATGATGCTAAA; Foxp2: AGCTCCTTTGCCTTCTCCACTCTT and AGGGCAATGAAGCCAGTCTGTACT.
Statistical Analysis
Graphpad Prism 5 software was used to calculate Kaplan-Meier survival curves and bar graph analyses. Data are presented as mean ± SEM.
References
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Bigner, S. H., Mark, J. & Bigner, D. D. Cytogenetics of human brain tumors. Cancer genetics and cytogenetics 47, 141–154 (1990).
Hahn, P. J. Molecular biology of double-minute chromosomes. BioEssays: news and reviews in molecular, cellular and developmental biology 15, 477–484 (1993).
Vogt, N. et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci U S A 101, 11368–11373 (2004).
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011).
Black, J. C. et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154, 541–555 (2013).
Lengauer, C., Kinzler, K. W. & Vogelstein, B. Genetic instabilities in human cancers. Nature 396, 643–649 (1998).
Attolini, C. S. et al. A mathematical framework to determine the temporal sequence of somatic genetic events in cancer. Proc Natl Acad Sci U S A 107, 17604–17609 (2010).
Hermanson, M. et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52, 3213–3219 (1992).
Guha, A., Dashner, K., Black, P. M., Wagner, J. A. & Stiles, C. D. Expression of PDGF and PDGF receptors in human astrocytoma operation specimens supports the existence of an autocrine loop. Int J Cancer 60, 168–173 (1995).
Fleming, T. P. et al. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res 52, 4550–4553 (1992).
Nister, M. et al. Expression of messenger RNAs for platelet-derived growth factor and transforming growth factor-alpha and their receptors in human malignant glioma cell lines. Cancer Res 48, 3910–3918 (1988).
Brennan, C. et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One 4, e7752 (2009).
Ozawa, T. et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev 24, 2205–2218 (2010).
Alentorn, A. et al. Prevalence, clinico-pathological value and co-occurrence of PDGFRA abnormalities in diffuse gliomas. Neuro Oncol 14, 1393–1403 (2012).
Calzolari, F. & Malatesta, P. Recent insights into PDGF-Induced gliomagenesis. Brain Pathol 20, 527–538 (2010).
Hambardzumyan, D., Parada, L. F., Holland, E. C. & Charest, A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia 59, 1155–1168 (2011).
Olson, L. E. & Soriano, P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16, 303–313 (2009).
Pringle, N. P., Mudhar, H. S., Collarini, E. J. & Richardson, W. D. PDGF receptors in the rat CNS: during late neurogenesis, PDGF alpha-receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development 115, 535–551 (1992).
Heinrich, M. C. et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299, 708–710 (2003).
Kang, S. H., Fukaya, M., Yang, J. K., Rothstein, J. D. & Bergles, D. E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681 (2010).
Liu, C. et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221 (2011).
Snuderl, M. et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20, 810–817 (2011).
Lundberg, G. et al. Binomial mitotic segregation of MYCN-carrying double minutes in neuroblastoma illustrates the role of randomness in oncogene amplification. PLoS One 3, e3099 (2008).
Chen, J., McKay, R. M. & Parada, L. F. Malignant glioma: lessons from genomics, mouse models and stem cells. Cell 149, 36–47 (2012).
Siebzehnrubl, F. A., Reynolds, B. A., Vescovi, A., Steindler, D. A. & Deleyrolle, L. P. The origins of glioma: E Pluribus Unum? Glia 59, 1135–1147 (2011).
Lindberg, N., Kastemar, M., Olofsson, T., Smits, A. & Uhrbom, L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene 28, 2266–2275 (2009).
Lindberg, N. et al. Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. J Neurosci 34, 14644–14651 (2014).
Chojnacki, A., Mak, G. & Weiss, S. PDGFRalpha expression distinguishes GFAP-expressing neural stem cells from PDGF-responsive neural precursors in the adult periventricular area. J Neurosci 31, 9503–9512 (2011).
Su, E. J. et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med 14, 731–737 (2008).
Jackson, E. L. et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51, 187–199 (2006).
Holland, E. C., Hively, W. P., DePinho, R. A. & Varmus, H. E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 12, 3675–3685 (1998).
Zhu, H. et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A 106, 2712–2716 (2009).
Little, S. E. et al. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res 72, 1614–1620 (2012).
Szerlip, N. J. et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc Natl Acad Sci U S A 109, 3041–3046 (2012).
Huse, J. T., Phillips, H. S. & Brennan, C. W. Molecular subclassification of diffuse gliomas: seeing order in the chaos. Glia 59, 1190–1199 (2011).
Chow, L. M. et al. Cooperativity within and among Pten, p53 and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell 19, 305–316 (2011).
Ozawa, T. et al. Most Human Non-GCIMP Glioblastoma Subtypes Evolve from a Common Proneural-like Precursor Glioma. Cancer Cell 26, 288–300 (2014).
Nathanson, D. A. et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76 (2014).
Sanborn, J. Z. et al. Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res 73, 6036–6045 (2013).
Tronche, F. et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23, 99–103 (1999).
Zhuo, L., Theis, M., Alvarez-Maya, I., Brenner, M., Willecke, K. & Messing, A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31, 85–94 (2001).
Serrano, M., Lee, H., Chin, L., Cordon-Cardo, C., Beach, D. & DePinho, R. A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37 (1996).
Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21, 70–71 (1999).
Rietze, R. L. & Reynolds, B. A. Neural stem cell isolation and characterization. Methods Enzymol 419, 3–23 (2006).
Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28, 264–278 (2008).
Acknowledgements
This work was supported by funds from the Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, to H.Z. and R.H.F. by development funds from the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, to R.H.F. and P.S. and by grants R01HD24875 and R37HD25326 from the NICHD to P.S.
Author information
Authors and Affiliations
Contributions
H.Z., L.E.O., P.S. and R.H.F. designed research; R.F., Y.H., J.T., M.J., L.E.O. and R.H.F. conducted experiments; H.Z., V.N., N.T., L.E.O., P.S. and R.H.F. analyzed data; H.Z. and R.H.F. wrote the paper. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zou, H., Feng, R., Huang, Y. et al. Double minute amplification of mutant PDGF receptor α in a mouse glioma model. Sci Rep 5, 8468 (2015). https://doi.org/10.1038/srep08468
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08468
This article is cited by
-
EPHA2 mediates PDGFA activity and functions together with PDGFRA as prognostic marker and therapeutic target in glioblastoma
Signal Transduction and Targeted Therapy (2022)
-
Molecular characterization of cell-free eccDNAs in human plasma
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
