
Abstract
The RAST (Rapid Annotation using Subsystem Technology) annotation engine was built in 2008 to annotate bacterial and archaeal genomes. It works by offering a standard software pipeline for identifying genomic features (i.e., protein-encoding genes and RNA) and annotating their functions. Recently, in order to make RAST a more useful research tool and to keep pace with advancements in bioinformatics, it has become desirable to build a version of RAST that is both customizable and extensible. In this paper, we describe the RAST tool kit (RASTtk), a modular version of RAST that enables researchers to build custom annotation pipelines. RASTtk offers a choice of software for identifying and annotating genomic features as well as the ability to add custom features to an annotation job. RASTtk also accommodates the batch submission of genomes and the ability to customize annotation protocols for batch submissions. This is the first major software restructuring of RAST since its inception.
Similar content being viewed by others

Introduction
The last two decades of research have brought vast changes to the field of genomics. The sequencing of a genome and the subsequent annotation of gene functions that were originally performed by teams of researchers expending thousands of man-hours of labor has become a standard laboratory technique that can be performed by a single person in one day. As sequencing technology has advanced and the cost has dropped, the number of genomes being deposited into the public databases has outpaced Moore's Law1,2. This has shifted the bottlenecks in genomic analysis from the sequencing per se to the tools that are used for annotation and genomic analysis.
In 2008, the RAST server (Rapid Annotation using Subsystem Technology) was developed to annotate microbial genomes3,4. It works by projecting manually curated gene annotations from the SEED database onto newly submitted genomes5,6,7. The key to the consistency and accuracy of the RAST algorithm has been the carefully structured annotation data in the SEED, which are organized into subsystems (sets of logically related functional roles)5. As a result, RAST has become one of the most popular sources for consistent and accurate annotations for microbial genomes. The RAST community currently consists of ~10,000 active users who have contributed an average of 1,170 microbial genomes per week in the last year. It is also being used as the foundation for maintaining consistency for automated metabolic modeling in the ModelSEED8 and KBase (kbase.us) and for comparative genomics in the bacterial pathogen database, PATRIC9,10.
RAST and other annotation engines encapsulate software for identifying and annotating specific genomic features into a standard annotation pipeline11,12,13,14,15,16. This approach has several advantages including offering speed, convenience and consistency to the user. In order to annotate with RAST, users submit their contigs to the server where the computation is performed. This frees users from having to download and install multiple programs, or to perform intensive computations. However, despite these advantages, this approach also has limitations. For instance, the default pipeline may not always be the best choice for a given genome. It is also difficult for researchers to customize annotation pipelines by choosing different tools and adding their own features and annotations. Until recently, it has also been difficult to submit batches of genomes and demand has been increasing for a version of RAST that can accommodate custom batch submissions.
In this paper, we describe a new modular implementation of RAST, which we call the RAST tool kit (RASTtk). RASTtk allows users to build their own annotation pipelines with a choice of gene calling algorithms, annotation scripts and output formats. It also provides a framework for users to add features and annotations to a processed RAST job. RASTtk can handle batch submissions of genomes, as well as the batch submission with custom annotation pipelines. RASTtk can be used on both the RAST website (http://rast.nmpdr.org) and the command line.
Results and Discussion
Accessing RASTtk
In order to make RAST more flexible and to keep pace with advancements that are being made in bioinformatics, we have separated the individual steps of the RAST annotation pipeline to provide a version that is modular, extensible and customizable.
RASTtk exists as a set of advanced options on the RAST web server (http://rast.nmpdr.org) (Figure 1). It has also been made available as a set of stand-alone scripts in the Interactive Remote Invocation Service (IRIS) environment (http://iris.theseed.org/). IRIS is a web application that functions like a command line window. Through IRIS, users can use the latest RASTtk scripts to build and compare custom annotation pipelines. In addition, RASTtk scripts can be installed and run locally using the RASTtk.app DMG (Mac only) (https://github.com/TheSEED/RASTtk-Distribution/releases/). Individual scripts are also available through the KBase GitHub page (https://github.com/kbase/genome_annotation). Tutorials for using the RASTtk scripts can be found on the SEED website (http://tutorial.theseed.org).

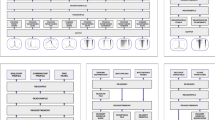
RASTtk options that are available on the RAST website (http://rast.nmpdr.org).
A table of options is displayed when the user selects the RASTtk annotation scheme and clicks the checkbox for “Customize”. Individual steps can be turned off and on using the check boxes. Parameters and conditions can be changed or added as needed. Dragging and dropping table rows will change the order of the steps.
The RASTtk Default Pipeline
During an annotation job, the data from individual scripts must be collected and integrated into a coherent picture of the genome as a whole. The abstract layout of a RASTtk pipeline is described below:
-
1
An initial tool converts a set of contigs (with a minimal amount of metadata) into a special file format called a Genome Typed Object (GTO). A GTO is a Java Script Object Notation (JSON) formatted file that is human-readable and allows for easy exchange of data objects [www.json.org].
-
2
Each step in the pipeline transforms an input GTO into an enhanced GTO. For example, calling genes with Prodigal uses the command “rast-call-features-CDS-prodigal” to add gene calls to an input GTO, which produces an expanded GTO (see Table 1 for a list of the commands implementing the current set of supported transformation tools).
Table 1 Characteristics of the RASTtk scripts -
3
A user can export data from a GTO. Generally, the final product of a sequence of transformation commands would be used to export a tab-delimited spreadsheet or GenBank entry17; numerous alternative export formats are also supported.
We offer a default pipeline for RASTtk, which represents our recommendation for a rapid and accurate annotation workflow. This workflow differs slightly from the “classic” RAST pipeline (Figure 2). For instance, we have added Prodigal18 as an additional gene caller because of its improved accuracy with short genes and start positions and because it is more robust to differences in G+C content12. We have also rewritten several of the core RAST algorithms, including the tools that find rRNA genes and resolve overlapping features. We have included a new version of the k-mer-based annotation algorithm19 and scripts that find repeat regions, CRISPRs, insertion sequences and Streptococcus repeats.

The RAST workflow.
Each individual step is bounded by a box and steps are connected by arrows. New RASTtk steps are indicated by red boxes and arrows. Improvements in the original steps are indicated in red text. Steps that are no longer part of the RASTtk pathway are indicated by gray arrows.
Calling ribosomal RNAs
Many tools exist for finding and curating collections of rRNAs13,20, but we needed a program that is simple, lightweight and fast. The current RAST rRNA finder is a custom script that uses hand-curated and phylogenetically diverse set of representative sequences of the 23S (currently 81 representatives), 16S (currently 120 representatives) and 5S (currently 292 representatives) rRNAs. These sets represent the diversity of genomes in the SEED and have been curated for the correct endpoints. The rRNAs of a new genome are simply found using a BLASTN21 search against this curated set. This script also reports partial length matches because genome assemblies with incomplete rRNA operons have become common.
Calling tRNAs
In order to find the tRNAs, we currently use tRNAscan-SE, a tool that was written by Lowe and Eddy22. It uses a secondary structure based searching method to find the tRNA genes.
Calling large repeat regions
The annotation of genomic regions that have been acquired by horizontal gene transfer remains a major challenge to maintaining consistency and accuracy in RAST. Repeat regions are often indicative of horizontal gene transfer and are hallmarks of insertion sequences and other mobile elements. Because of this, we have added a new script to the default RASTtk pipeline that performs a BLASTN21 search of the genome against itself and reports any region occurring more than once with ≥ 95% nucleotide identity. These precomputed repeat regions can then be used for comparative analyses and as supporting data for more detailed annotation of mobile elements.
Calling seleno- and pyrrolysylproteins
Selenoproteins are widespread among the sequenced bacterial and archaeal genomes occurring in ~25% of the genomes in the CoreSEED (a collection of ~1000 highly curated diverse bacterial and archaeal genomes utilized by RAST). They contain the rare amino acid selenocysteine, which is incorporated at a UGA stop codon in frame23. In order to find these proteins, a hand-curated set of known selenoproteins is kept in a BLAST database21. When a match to a potential selenoprotein is found within a genome, it is then searched for the in-frame stop codon. If the stop is found, then the protein is annotated as a selenoprotein. Pyrrolysylproteins are less common among the currently sequenced genomes, occurring in ~1% of the sequenced bacterial and archaeal genomes in the CoreSEED. Similar to selenocysteine, pyrrolysine is incorporated at a UAG stop codon23. We search for pyrrolysylproteins using the same strategy.
Calling Streptococcus repeat elements
Streptococcus species contain small interspersed repeats that may modulate gene expression and can be used for epidemiological typing24,25. We have added tools created by Croucher et al. for finding these elements24. When the user specifies the metadata for the genome, these scripts will be run if the genus is Streptococcus. In the future, we anticipate adding other species-specific annotation tools and their conditional usage in RASTtk will mirror that of the Streptococcus repeat finder.
Calling CRISPR elements
CRISPRs, clustered regularly interspaced short palindromic repeats, are a special type of repeat region found in many bacterial and archaeal genomes. The CRISPR array contains spacer regions matching foreign DNA that are regularly spaced and bounded by repeat regions. The DNA of the spacer region is transcribed and used to interfere with incoming foreign DNA. Because of their importance in horizontal gene transfer and in biotechnology26,27, we have added a script to the RASTtk pipeline that finds CRISPR elements. This script works by using a Perl regular expression search [Wall, L., Christiansen, T. & Orwant, J. Programming perl. (O'Reilly Media, Inc., 2004)] to find recurring direct repeats of at least 24 nucleotides in length and spaced at regular intervals. The output of the script is three new feature types: the entire CRISPR array, the CRISPR spacer and the CRISPR repeat.
Calling genes
RASTtk offers the option to call the open reading frames with Glimmer328, GeneMarkS29 and Prodigal18. The original web-based version of RAST used Glimmer3 and our current default RASTtk pipeline uses both Prodigal and Glimmer3. The output of both programs is added to the GTO file and then an optimal set of calls is chosen in the overlap resolution step (described below).
Annotating proteins with k-mers
Historically, RAST has made heavy use of the FIGfam collection maintained within the SEED project6. FIGfams are protein families in which it is believed that all members of the same family share an identical function and were derived from a common ancestor (i.e., they are all isofunctional homologs). The original implementation of the k-mer-based assignment of function was based on the use of signature k-mers3,19. A signature k-mer is defined as an 8-mer amino acid sequence in which the vast majority (over 80%) of occurrences are found within FIGfams sharing a common function and that do not occur in any FIGfam with a different function. For example, a k-mer for which 93% of the occurrences within the FIGfam collection were in families implementing the function SSU ribosomal protein S13p (S18e) would be considered a “signature of function”. In this case, the signatures depend critically on the FIGfams.
Once we had modestly consistent collections of sequences in the SEED, we introduced a version of k-mer analysis that was not based on FIGfams. The notion of signature k-mer was modified to an 8-mer amino acid sequence in which the vast majority (over 80%) of occurrences were within sequences assigned an identical function. This version depends on the collection of protein sequences with consistent annotations. Currently, we base this version on a collection of about 1000 representative genomes present in a subset of the SEED called the CoreSEED (core.theseed.org). The CoreSEED represents our best attempt at annotation consistency and is currently the main focus of our manual annotation efforts.
The CoreSEED database attempts to provide the most consistent manual annotations for the core metabolic and house keeping functions in a relatively small and diverse set of the bacteria and archaea, whereas the PubSEED database (from which the FIGfam collection is generated) attempts to absorb new annotations from the academic community for many genomes. The default RASTtk workflow first searches against the limited number of more stable annotation-based k-mers from the CoreSEED and then if an annotation cannot be found it searches against the larger collection of FIGfam based k-mers from the PubSEED.
In addition to these two k-mer based gene-function assignment tools, there are also two analogous tools that use these k-mer sets to search for function-containing regions of DNA that do not require gene calls. They are useful for searching for genes in regions where calls may have been missed and for assessing functions in un-assembled sequence data.
Annotating proteins missed by k-mers
If no function can be found for a protein-encoding gene during the k-mer analysis, a final search that uses a combination of BLAT30 and BLASTP21 is performed against a set of non-redundant genus-specific protein databases for the organism's genus and, when available, closest relatives. If a matching protein is found with an e-value < = 1e-5 and a percent identity > = 50%, then the function from the protein in the database is assigned to the gene.
Resolving Overlapping Calls
After using different tools to call open reading frames and annotate features, we try to resolve the results into a coherent picture. To resolve overlapping features, we use a dynamic programming algorithm that resembles the scoring algorithm in Prodigal18. It works by scanning the genome and generating a score for each alternative combination of feature calls for tRNA, rRNA and protein-encoding genes. In general, for a given location on the contig, tRNA and rRNA receive a higher score than protein encoding genes and large overlaps receive a negative score based on the length of the overlap. Large gaps between genes also result in negative scores. After considering all of the combinations calls, the genomic arrangement with the highest score is chosen. User-defined features are exempt from consideration by this algorithm.
Additional Analysis Scripts
Customizing and updating an annotation job
Users can run custom analysis jobs locally on the command line or in IRIS and then add their own features to an annotation job. In order to input specialty features, the user must create a tab-delimited text file, which contains a unique identifier, the location, feature type and function. These are then added to the GTO file and can be exported in a variety of formats. Users can also update functions directly by providing a tab-delimited file with the identifier and the new function. The update is then logged in the GTO file.
Batch genome submissions
RASTtk supports the ability to upload a directory of GTO files either using IRIS or the RASTtk.app DMG. When a batch upload is performed, the entire directory is uploaded to the RAST server and placed into the queue. A job identifier is returned to the user and this is used to check the status of the job and to download the job when it is completed. A custom RASTtk pipeline can be invoked by adding a special JSON formatted workflow file along with the directory of genomes. For more information on using RASTtk in batch mode, please refer to the RASTtk tutorials (http://tutorial.theseed.org).
Calling prophage elements
In order to find potential prophage elements we have added PhiSpy31. PhiSpy uses a combination of several independent heuristic methods to identify regions in the genome, which may be derived from phages or mobile elements.
Finding insertion sequences
We have added a new tool that uses a reference set of end sequences and transposase proteins from the SEED and ISfinder databases3,32 to search the genome for IS elements. Matches are found by using a combination of BLASTN for the end regions and BLASTX for the proteins21.
Identifying special gene sets
The PATRIC project is an integration of data and tools for studying bacterial pathogens. In order for RASTtk to support PATRIC, it was necessary to improve the identification of genes relating to virulence and drug development33. RASTtk now offers an analysis script for this purpose. It searches against custom BLAST databases that have been built from ARDB34 and CARD35 for finding potential antibiotic resistance genes; DrugBank36 and TTD37 for finding potential drug targets; VFDB38, Victors39 and the PATRIC virulence factors10,33 for finding potential virulence factors; and the human reference genome sequence40 for finding potential human homologs, which is an important step in drug screening analyses.
Future Directions
RASTtk enables users to optimize and customize the annotation steps for a given genome and to apply these pipelines to sets of genomes as customized batch submissions. The modularity of RASTtk also makes it much easier to develop and incorporate software for improving genome annotations and we anticipate adding tools to RASTtk as they become needed. We also expect the utilization of RASTtk to result in the community development of annotation pipelines aimed at solving more specialized annotation problems, such as more accurate gene calling in prophages and eukaryotes. We also anticipate providing specialty scripts that improve the accuracy for gene families that are currently difficult to annotate, such as pathogenicity-related genes, transporters and mobile elements.
References
Shendure, J. & Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 26, 1135–1145 (2008).
Hayden, E. C. Technology: the $1,000 genome. Nature. 507, 294–295 (2014).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. gkt1226 (2013).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
Overbeek, R. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 (2005).
Meyer, F., Overbeek, R. & Rodriguez, A. FIGfams: yet another set of protein families. Nucleic Acids Res. 37, 6643–6654 (2009).
Davis, J. J., Olsen, G. J., Overbeek, R., Vonstein, V. & Xia, F. In search of genome annotation consistency: solid gene clusters and how to use them. 3 Biotech. 1–5 (2013).
Henry, C. S. et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 (2010).
Gillespie, J. J. et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 79, 4286–4298 (2011).
Wattam, A. R. et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. gkt1099 (2013).
Almeida, L. G. et al. A system for automated bacterial (genome) integrated annotation—SABIA. Bioinformatics. 20, 2832–2833 (2004).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. btu153 (2014).
Tatusova T, D. M. & Badretdin, A. et al. [Prokaryotic Genome Annotation Pipeline]. The NCBI Handbook [Internet] 2nd ed. 1–23 National Center for Biotechnology Information, Bethesda, MD USA, 2013.
Mavromatis, K. et al. The DOE-JGI Standard operating procedure for the annotations of microbial genomes. Stand. Genomic Sci. 1, 63 (2009).
Utley, J. M. R-FAP: Rapid Functional Annotation of Prokaryotes Using Taxon-specific Pan-genomes and 10-mer Peptides. Masters thesis, University of Tennessee, 2014.
Markowitz, V. M. et al. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34, D344–D348 (2006).
Benson, D. A. et al. GenBank. Nucleic Acids Res. gks1195 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 11, 119 (2010).
Edwards, R. A. et al. Real Time Metagenomics: Using k-mers to annotate metagenomes. Bioinformatics. 28, 3316–3317 (2012).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics. 10, 421 (2009).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 0955–0964 (1997).
Cobucci-Ponzano, B., Rossi, M. & Moracci, M. Translational recoding in archaea. Extremophiles. 16, 793–803 (2012).
Croucher, N. J., Vernikos, G. S., Parkhill, J. & Bentley, S. D. Identification, variation and transcription of pneumococcal repeat sequences. BMC Genomics. 12, 120 (2011).
van Belkum, A., Sluijuter, M., de Groot, R., Verbrugh, H. & Hermans, P. Novel BOX repeat PCR assay for high-resolution typing of Streptococcus pneumoniae strains. J. Clin. Microbiol. 34, 1176–1179 (1996).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science. 339, 823–826 (2013).
Sorek, R., Kunin, V. & Hugenholtz, P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 6, 181–186 (2008).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 23, 673–679 (2007).
Borodovsky, M. & Lomsadze, A. Gene identification in prokaryotic genomes, phages, metagenomes and EST sequences with GeneMarkS suite. Curr. Protoc. Bioinformatics. 4.5. 1–4.5. 17 (2011).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Akhter, S., Aziz, R. K. & Edwards, R. A. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity-and composition-based strategies. Nucleic Acids Res. 40, e126–e126 (2012).
Siguier, P., Pérochon, J., Lestrade, L., Mahillon, J. & Chandler, M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36 (2006).
Mao, C. et al. Curation, integration and visualization of bacterial virulence factors in PATRIC. Bioinformatics. btu631 (2014).
Liu, B. & Pop, M. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447 (2009).
McArthur, A. G. et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357 (2013).
Law, V. et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097 (2014).
Qin, C. et al. Therapeutic target database update 2014: a resource for targeted therapeutics. Nucleic Acids Res. 42, D1118–D1123 (2014).
Chen, L., Xiong, Z., Sun, L., Yang, J. & Jin, Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. gkr989 (2011).
Xiang, Z., Tian, Y. & He, Y. PHIDIAS: a pathogen-host interaction data integration and analysis system. Genome Biol. 8, R150 (2007).
Church, D. M. et al. Modernizing reference genome assemblies. PLoS Biol. 9, e1001091 (2011).
Acknowledgements
We thank Emily Dietrich for her helpful comments. This work was supported by the United States National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Service [Contract No. HHSN272201400027C]; the United States Department of Energy [DE-AC02-06CH11357], as part of the DOE Systems Biology Knowledgebase; R.A.E. was supported by United States National Science Foundation Grants grants II-EN: Computational Enhancement of Analytical Metagenomics Systems CNS-1305112 and Experimental and Computational Determination of Microbial Genotypes and Phenotypes MCB-1330800; and G.J.O. was supported by the National Aeronautics and Space Administration through the NASA Astrobiology Institute under Cooperative Agreement No. NNA13AA91A issued through the Science Mission Directorate. United States Department of Energy: National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Contributions
T.B., J.J.D., T.D., R.A.E., S.G., G.J.O., R.O.l., R.O.v., B.P., G.D.P., M.S., J.A.T., R.S., V.V., A.R.W. and F.X. designed the project, built the RASTtk software and contribute to the maintenance of the RAST tool kit. J.J.D. and R.O.v. wrote this manuscript. All authors have reviewed this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Brettin, T., Davis, J., Disz, T. et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci Rep 5, 8365 (2015). https://doi.org/10.1038/srep08365
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08365
This article is cited by
-
Organic matter degradation in the deep, sulfidic waters of the Black Sea: insights into the ecophysiology of novel anaerobic bacteria
Microbiome (2024)
-
Genome characterisation and comparative analysis of Schaalia dentiphila sp. nov. and its subspecies, S. dentiphila subsp. denticola subsp. nov., from the human oral cavity
BMC Microbiology (2024)
-
Genetic characteristics of chromosomally integrated carbapenemase gene (blaNDM−1) in isolates of Proteus mirabilis
BMC Microbiology (2024)
-
Actinomycetospora termitidis sp. nov., an insect-derived actinomycete isolated from termite (Odontotermes formosanus)
The Journal of Antibiotics (2024)
-
Emergence of multidrug resistant, ctx negative seventh pandemic Vibrio cholerae O1 El Tor sequence type (ST) 69 in coastal water of Kerala, India
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
