
Abstract
Glioma is a rare, but highly fatal, cancer that accounts for the majority of malignant primary brain tumors. Inherited predisposition to glioma has been consistently observed within non-syndromic families. Our previous studies, which involved non-parametric and parametric linkage analyses, both yielded significant linkage peaks on chromosome 17q. Here, we use data from next generation and Sanger sequencing to identify familial glioma candidate genes and variants on chromosome 17q for further investigation. We applied a filtering schema to narrow the original list of 4830 annotated variants down to 21 very rare (<0.1% frequency), non-synonymous variants. Our findings implicate the MYO19 and KIF18B genes and rare variants in SPAG9 and RUNDC1 as candidates worthy of further investigation. Burden testing and functional studies are planned.
Similar content being viewed by others

Introduction
Gliomas comprise approximately 75% of all malignant primary brain tumors (PBTs) and account for an estimated 4% of cancer deaths in the United States1,2,3,4. Glioblastoma multiforme (GBM) is the most common type of glioma, constituting nearly 65% of cases, with an incidence rate of 2–3 per 100,000 in the United States and Europe. The 5-year survival for individuals with GBM is only about 10% and median survival time is an estimated 12–14 months.
Despite decades of research, there are few established risk factors for glioma5. A number of candidate gene and genome wide association studies have been conducted6,7,8,9,10 and have, thus far, revealed seven low-penetrant susceptibility loci associated with sporadic glioma formation8. With regard to familial glioma, known single gene disorders, such as neurofibromatosis, tuberous sclerosis and Li-Fraumeni and Turcot's syndromes, predispose patients to glioma formation, but cannot explain more than a minute proportion of cases11. Therefore, the factors responsible for first-degree relatives of glioma patients having approximately twice the risk of glioma formation compared to unrelated individuals remains unclear12.
Because the genetic basis of familial glioma remains enigmatic, the “Genetic Epidemiology of Glioma International Consortium” (Gliogene Consortium) was formed in 2006 to recruit families affected by glioma in 14 different institutions across five countries11,13,14. This consortium has provided an unprecedented opportunity to further our understanding of the heritability of this rare, though highly fatal, condition, with the ultimate goal of uncovering enough information about glioma susceptibility to allow for the screening and genetic counseling of high-risk individuals and families. Our previous studies, which involved non-parametric and parametric linkage analyses, both yielded significant linkage peaks only on chromosome 17q (parametric linkage score: 3.1, nonparametric linkage score: 3.39)13,15. Because of the concordance between these prior findings, we conducted targeted sequencing focused on this region of chromosome 17q, with the aim of identifying the variant(s) or gene(s) that could explain linkage across this region to familial glioma. We, additionally, characterized deleterious rare variants within this chromosomal region among these glioma families.
Results
The linkage region was defined as the 1.7 LOD drop region from linkage peaks, which spans bases 34,355,567-52,135,011 and 54,612,056-61,596,548 (GRCh37 coordinates) on chromosome 17 (solid bars in Fig. 1).

Combined results of parametric and non-parametric linkage analysis on Gliogene families.
The only significant peaks are on chromosome 17q. All coordinates are based on human genome version 19 sequence (GRCh37). Solid bars represent the linkage locus, defined as the 1.7 LOD drop region.
A total of 203 individuals from 23 families were successfully sequenced (33 affected, 170 unaffected) to an average depth of coverage of 95 fold in the target regions. Of these families, 20 had at least one affected individual sequenced (probands of three families failed sequencing and were subsequently excluded from additional analysis). Information on the demographics of affected individuals, whether sequencing was completed and the relationship between affected individuals in each family is summarized in Table 1. Affected individuals were not sequenced if they were deceased prior to study initiation, or if a specimen was otherwise unobtainable.
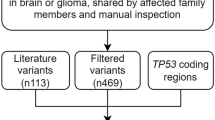
After alignment and variant calling, a total of 4,830 variants were annotated. After removing common variants, variants mapping outside our selected target regions, variants only present in unaffected individuals and variants not segregating among affected individuals in the same family, there were 539 remaining variants, which were subsequently submitted for Sanger sequencing verification for the affected individuals (Fig. 2A). All of the variants in our final list (Table 2) were located within the linkage region.

Chromosome 17q variant filtering schema.
(a) Flowchart depicts the initial set of variant filters used to narrow the list of variants for Sanger sequencing. “Targeted region” and “Linked locus” filters were waived for variants in Cancer Gene Census53 genes. (b) Flowchart depicts the quality check process for Sanger-confirmed variants and the post Sanger sequencing set of variant filters applied to arrive at the final list of 21 candidate variants.
Sanger sequencing
Out of 539 variants interrogated by Sanger sequencing, some failed initial design and quality checks, including several attempts at successful primer design, in part due to the location of the variants within high-repeat or low-complexity regions or within duplicated regions. A total of 278 variants (51.6%) were validated in at least in a subset of affected individuals (Fig. 2B). Reads not meeting the aforementioned quality control criteria were removed, leaving 186 variants (66.9%) that were fully verified by Sanger sequencing. Our final filtering criteria were then applied (Fig. 2B). A final list of 21 candidate variants, all missense mutations, was obtained (Table 2).
Candidate variants
The 21 candidate variants on our final list are each private to individual families (Table 2). There were 15 candidate variants present in more than one affected individual per family (“2/2” affected ratio in Table 2) and the remaining six variants were in families with only one sequenced affected individual. Of the 15 shared variants, three variants are novel, never having been reported in a publicly available variant database (dbSNP, ESP, 1000 Genome Project). These include two missense mutations in RSAD1 and MYO19 in both affected and 3 out of 8 unaffected members of Family J and a missense mutation in G6PC in both affected and 0 out of 5 unaffected members of Family A.
Although each specific variant on our final list was private to an individual family, there were two genes for which more than one family had variants that made the final list. MYO19 was implicated in Families J and O and KIF18B was implicated in Families G and B (Table 2).
Of the 21 candidate variants identified by our filtering schema, 19 had scaled C-scores above 10, indicating that these variants are predicted to be in the top 10% of the most deleterious possible substitutions in the human genome. The highest scaled C-scores belonged to SPAG9 rs143491486 (C-score: 32.0), RUNDC1 rs61995866 (C-score: 28.2) and the variant in TTC25 (chr17:40091564 C>G; C-score: 25.9). In Family L, both sequenced affected individuals and 3 out of 4 sequenced unaffected individuals had the SPAG9 rs143491486 variant. However, 2 of the 3 unaffected individuals who had this SPAG9 variant were children (Fig. 3a). The abbreviated CADD output is provided as supplementary information.

(a) SPAG9(L) and (b) RUNDC1(C) mutations in familial glioma pedigrees. Individuals with glioma are shown as filled. Individuals with other cancers are shown as half-filled. Disease and age in years (y) at first diagnosis is given underneath the symbol, current age or age at death (+) above it. Glioma type is shown (AA, anaplastic astroctyoma; Astro, astrocytoma; GBM, glioblastoma multiforme). Other cancers in the pedigree are shown (OvC, ovarian cancer; Mel, melanoma;TesC, testicular cancer; Lym, lymphoma). Carriers of SPAG9 (a) and RUNDC1 (b) mutations are shown with their specific mutation, whereas individuals who tested negative for the mutation in the specific pedigree are depicted as wild-type (WT).
Discussion
In this study, we attempted to identify variants on targeted regions of chromosome 17q that could explain linkage across familial glioma cases. We also sought to describe the distribution of potentially deleterious rare variants within participating glioma families. As in any NGS study, we obtained a large list of variants and had to devise a filtering and verification strategy to refine our list for inclusion of only the most relevant variants (Fig. 2). Although we did not detect a variant (or gene) that could explain linkage across all participating families, we did identify several very rare or novel missense variants (many of which are predicted to be in the top 10% of the most deleterious substitutions possible in the human genome) that segregated in sequenced affected members of individual families. In complex diseases such as glioma, the importance of identifying deleterious rare variants (e.g., private mutations) should not be underestimated, as such variants may pinpoint genes that could be important to disease pathogenesis. Furthermore, despite being infrequent in the general population, the identification of these rare variants can highlight genes or genomic regions in which a series of less deleterious and more common mutations may interact to cumulatively increase disease risk in a larger population.
Two interesting candidate genes identified in this study are MYO19 and KIF18B, which were the only genes that were implicated in more than one family. MYO19 codes for a myosin that is involved in mitochondrial motility16. The MYO19 gene has not been well-studied in relation to carcinogenesis; however, one study has implied that it may act as a fusion gene in breast cancer tissues17. KIF18B encodes a protein product that regulates microtubule dynamics (i.e., microtubule length) and thus, plays an important role in cell division through its involvement in mitotic spindle assembly18,19,20. KIF18B has been shown to be overexpressed in hepatocellular carcinoma and some evidence suggests that the expression of this gene may also be deregulated in several other types of tumor tissues20,21. Both of these genes warrant additional examination in future etiologic studies.
A promising candidate variant identified in our study, SPAG9 rs143491486 (p.Ser269Leu), is predicted to be among the top 0.1% of the most deleterious variants possible in the human genome (Table 2, Family L), based on its Combined Annotation-Dependent Depletion22 scaled C-score of 32. The product of this gene, sperm-associated antigen 9 (SPAG9), is a member of a scaffolding protein family that helps MAP-kinases bind with their transcription factors to activate specific signal transduction pathways23,24,25. SPAG9 is actually a putative oncoprotein that has been implicated in prostate, breast, hepatocellular, thyroid, cervical, lung, bladder and endometrial cancers23,24,26,27,28,29,30,31,32,33,34. Recently, Yi et al. showed that SPAG9 was differentially overexpressed in human astrocytomas, compared to normal astrocytes and that SPAG9 expression levels were positively correlated with tumor grade (p<0.001)24. Interestingly, in our study, one of the two sequenced affected individuals carrying the SPAG9 variant in Family L had a Grade II astrocytoma (Fig. 3a). Although the exact mechanism through which this particular SPAG9 variant may be involved in gliomagenesis is unknown, it has been suggested that the SPAG9 protein may influence cell invasion by upregulating the expression of MMP9 (matrix metallopeptidase 9, a.k.a. gelatinase-B), which, in turn, has been shown to be involved in the neovascularization of malignant gliomas35,36.
The non-synonymous SPAG9 variant identified in our study contributes to existing evidence that this gene may play an important role in glioma susceptibility. This variant is predicted to be protein damaging by SIFT (SIFT score: 0; http://sift.jcvi.org/)37 and PolyPhen (Polymorphism Phenotyping score: 0.997)38 and is at a location that is highly evolutionarily conserved. Ascertainment of whether currently unaffected members of Family L who carry this variant develop glioma in the future will be extremely important, as the unaffected individuals carrying this variant were under the age of 20 and may develop glioma later in life (Fig. 3a).
There is little known about the variant with the next highest C-score, RUNDC1 rs61995866 (p.Glu386Gln). This variant is predicted to be in the 1% most deleterious of all possible substitutions in the human genome. The RUNDC1 protein has been shown by a high-throughput RNA interference-based screening study to be a p53 regulator39,40. Interference with this gene resulted in increased p53 transcription, thus indicating that the RUNDC1 protein may be a p53 antagonist39. However, this variant has not previously been studied in relation to familial glioma. Both sequenced affected individuals in Family C carried this mutation, as did two of four sequenced unaffected individuals (implying incomplete penetrance) [Fig. 3b]. However, one of the two unaffected individuals carrying the RUNDC1 variant was only in her early 20s at last contact.
Even the variants on our final list that have lower C-scores may provide interesting candidates for further investigation. For example, the candidate gene with the highest number of somatic non-silent mutations in The Cancer Genome Atlas (TCGA) low-grade glioma database is G6PC with three tumor samples containing missense somatic variants, followed by one tumor sample for each of MYO19, GHDC ¸ and KRT27. Looking at the combined low-grade glioma and glioblastoma multiforme TCGA data, G6PC remains the gene with the highest number of somatic non-silent mutations (four tumor samples) followed by MYO19 (three tumor samples) and KIF18B (two tumor samples). This was based on an analysis of 279 low-grade glioma and 268 glioblastoma multiforme tumor samples from the TCGA project as reported by the International Cancer Genome Consortium (https://dcc.icgc.org/).
Targeted deletion of G6PC in mice leads to hepatic tumorigenesis41 and this gene is expressed at comparable levels in the central nervous system according to the Cancer Genome Anatomy Project42. Along with the finding that both affected (with low grade glioma) and no unaffected members of Family A carried the G6PC missense variant in their germline (scaled C-score: 14.5), this information makes the G6PC variant another interesting candidate for future study.
It is also noteworthy that four different keratin genes were implicated in our final list (Table 2, Families C, H, K and J). Overexpression of cytokeratins has been reported in some cancers43,44,45, but little is known about the potential role of these genes in gliomagenesis. There is a cluster of keratin-related genes or pseudogenes on chromosome 17q,45 thereby obscuring whether these findings may truly be biologically relevant or simply represent an artifact of our loci of interest.
Glioma is a complex disease where multiple loci are likely to be important for disease development within a family. In order to maximize resources, the regions targeted for sequencing in our study were chosen strategically to ensure comprehensive coverage of what were likely to be the most pertinent linkage regions. However, within our sequencing targets, there are numerous regions that are notoriously difficult to sequence via the NGS platform and Sanger sequencing, possibly due to genomic duplications or high repeat content. Additionally, we acknowledge that other loci, outside of the regions covered by this study, have previously been linked to familial glioma46 (although no causative variants in these regions have been definitively established). For example, in our other analyses, we have found that two Gliogene families (D and R) had TP53 mutations that may help explain their increased glioma risk47. Interestingly, neither of these families had chromosome 17q variants that met our final filtering criteria in the current study.
One of the challenging aspects of studying diseases with high mortality, such as glioma, is that there are very few families for which we can obtain biologic samples from more than one or two affected family members, which limits the possibility of performing segregation analyses of rare variants. Only about 5% of glioma patients have a family history of glioma14 and in 83% of glioma families, there are only two glioma patients in the family11, indicating a complex disease with low penetrance of causal variants. Unfortunately, we were unable to detect a variant that could explain linkage across all participating families in this study. Because there are no established filtering strategies for studies such as this one, we had to develop the filtering schema presented here to maximize the potential of finding such variants, while simultaneously excluding variants common among healthy populations.
Despite these limitations, the findings of this study may lend a substantive amount of insight into genes involved in gliomagenesis and future studies should evaluate the candidate genes/regions identified here for the presence of more common/less deleterious variants that may cumulatively impact familial glioma risk in a larger population. Overall, our study complements the previous research conducted by the Gliogene Consortium on the enigmatic factors contributing to familial glioma risk. The importance of the chromosome 17q region is becoming increasingly clear, though several additional studies are necessary before definitively conclusions can be drawn. We are currently in the process of obtaining copy number variation (CNV) data for a subset of the Gliogene families. The CNV data will be examined in the context of what is currently known about factors that predict familial glioma susceptibility in the hopes that enough information can be amassed from the Gliogene Consortium's series of studies to be able to differentiate and provide genetic counseling to high-risk families and individuals.
Methods
Study Population
Families recruited for participation in the Gliogene Consortium had at least two biologically related members (83% of families with two and 17% with three or more affected family members) who were diagnosed with a histologically-confirmed glioma11. The study population and recruitment scheme have been described in detail elsewhere11,13,14,15. Briefly, for the analyses presented here, the glioma families (n = 23) contributing most to the linkage peaks identified in Shete et al. (2011)13 and Sun et al. (2012)15 were selected for genomic sequencing.
Sample collection
Blood or saliva samples from glioma family members were obtained under written informed consent at each Gliogene Consortium recruitment center. DNA samples were prepared as previously described13. This study was approved by the Institutional Review Board of each Gliogene institution, including Baylor College of Medicine and was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants or their guardians.
Library construction
DNA samples were constructed into Illumina paired-end pre-capture libraries according to the manufacturer's protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC protocol (Illumina Barcoded Paired-End_Capture_Library_Preparation). Libraries were prepared using Beckman robotic workstations (Biomek NXp and FXp models). Briefly, 1 ug of DNA was sheared into fragments of approximately 300–400 base pairs with the Covaris E210 system followed end-repair, A-tailing and ligation of the Illumina multiplexing PE adaptors. Pre-capture ligation-mediated PCR (LM-PCR) was performed for 6–8 cycles of amplification using the 2X SOLiD Library High Fidelity Amplification Mix (a custom product manufactured by Invitrogen). Purification was performed with Agencourt AMPure XP beads after enzymatic reactions and following the final purification, quantification and size distribution of the pre-capture LM-PCR product was determined using the LabChip GX electrophoresis system (PerkinElmer).
Custom capture design
The DNA capture reagent was designed to target all coding exons of all genes, the UTRs of cancer related genes, miRNA binding sites in all 3′ UTRs, transcription factor binding sites, miRNA and small nucleolar RNA within the chromosome 17 linkage peak region.
The pre-capture libraries were pooled as a 46-plex (totaling 1 ug per pool) and hybridized in solution to the custom Gliogene capture reagent (1.6 Mb, NimbleGen) according to the manufacturer's protocol (NimbleGen SeqCap EZ Exome Library SR User's Guide) with minor revisions. Human COT1 DNA was added into the hybridization to block repetitive genomic sequences, followed by post-capture LM-PCR amplification using the 2X SOLiD Library High Fidelity Amplification Mix with 14 cycles of amplification. After the final AMPure XP bead purification, quantity and size of the capture library was analyzed using the Agilent Bioanalyzer 2100 DNA Chip 7500. The efficiency of the capture was evaluated by performing a qPCR-based quality check on the enrichment level of four standard NimbleGen internal control loci. Successful enrichment of the capture libraries was estimated to range from a 6 to 9 of ΔCt value over the non-enriched samples.
Sequencing
Library templates were prepared for sequencing using Illumina's cBot cluster generation system with TruSeq PE Cluster Generation Kits. Briefly, these libraries were denatured and diluted in hybridization buffer in order to achieve a load density of ~800 K clusters/mm2. Each library pool was loaded in a single lane of a HiSeq flow cell, with 2% phiX control library spiked into the lane for run quality control. The sample libraries then underwent bridge amplification to form clonal clusters, followed by hybridization with the sequencing primer. The sequencing run was performed in paired-end mode using the Illumina HiSeq 2000 platform. Using the TruSeq SBS Kits, sequencing-by-synthesis reactions were extended for 101 cycles from each end, with an additional 7 cycles for the index read. With the sequencing runs yielding an average of ~838 Mb per sample, samples achieved an average of 86% of the targeted exome bases covered to a depth of 20X or greater.
Initial sequence analysis was performed using the HGSC Mercury analysis pipeline (https://www.hgsc.bcm.edu/content/mercury)48. In summary, the .bcl files produced on-instrument were first transferred into the HGSC analysis infrastructure by the HiSeq Real-time Analysis module. Mercury then ran the vendor's primary analysis software (CASAVA) to de-multiplex pooled samples and generate sequence reads and base-call confidence values (qualities), followed by the mapping of reads to the GRCh37 Human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows-Wheeler aligner (BWA, http://bio-bwa.sourceforge.net/)49. The resulting BAM (binary alignment/map)50 file underwent quality recalibration using GATK (http://www.broadinstitute.org/gatk/)51 and where necessary the merging of separate sequence-event BAMs into a single sample-level BAM. BAM sorting, duplicate read marking and realignment to improve insertion/deletion discovery all occur at this step.
Sanger validation
Mutation validation was performed with bidirectional sequencing of the selected sample sites. PCR reactions were prepared using 5 ng of genomic DNA, 0.4 µM oligonucleotide primers and 0.7X Qiagen Multiplex Master Mix (Cat. no. 206145) containing HotStar Taq, buffer and polymerase. Reactions were performed with and without QSOL PCR additive to enhance PCR and final sequence performance. Touchdown PCR was performed with the following parameters: 98°C for 5 min. and 10 cycles of 98°C for 30 sec., 72°C for 30 sec. (decreasing by 1°C per cycle) and 72°C for 1 min. The reaction then continued with 30 cycles of 98°C for 30 sec., 63°C for 30 sec. and 72°C for 1 min, followed by a final extension at 72°C for 5 min. The PCR products were purified with a 1:15 dilution of Exo-SAP, diluted by 0.6X and then cycle-sequenced for 25 cycles using a 1/64th dilution of BigDye Terminator v3.1 reaction mix (Applied Biosystems, Cat. No. 4337456). Finally, reactions were precipitated with ethanol, resuspended in 0.1 mM EDTA and analyzed on AB 3730xl sequencing instruments using the Rapid36 run module and 3xx base-caller. SNPs were identified using SNP Detector software and then visually validated with Consed.
Variant annotation and filtering
Variants were annotated for functional effect on protein and presence in variant databases (i.e., dbSNP), as well as known minor allele frequency using AnnoVar52. We developed a filtering schema (described below) to evaluate the full list of variants and reduce it to a subset of variants more likely to be etiologically relevant (i.e., very rare protein-altering candidate variants within the linkage region). We implemented this series of filtering strategies using scripts written in R (version 2.15.3).
Variants that met initial filtering criteria were verified by Sanger sequencing among affected individuals and an additional post-Sanger filtering strategy was then applied. Filtering was carried out in a stepwise fashion with verification of the excluded variants at each step to ensure that biologically significant variants were not being eliminated.
Initial Filtering Strategy
The borders of the linkage region were defined based on a 1.7 LOD drop region (~ 99% linkage confidence interval) from the linkage peaks identified in our previous two analyses13,15, which encompassed two adjacent regions on chromosome 17q. Variants located outside this linkage region were excluded (“Linked locus” filter), unless the variants outside the region were in genes found in the Cancer Gene Census (CGC)53. The CGC, which is maintained by Welcome Trust Sanger Institute, is a catalogue of variants that have previously been implicated to have a causal role in oncogenesis. At the time of this analysis, the following 23 genes on chromosome 17 were included in the CGC: TAF15, SUZ12, LASP1, CLTC, CDK12, NF1, RARA, BRCA1, DDX5, ETV4, MLLT6, ERBB2, COL1A1, BRIP1, HLF, CD79B, MSI2, PRKAR1A, MSF, ASPSCR1, CANT1, SRSF2, and ALO17.
Among non-CGC genes, variants that were not within 200 bases of the sequencing target regions were excluded (“Targeted Region” filter). Next, common variants, defined as those that had >5% frequency in the Exome Sequencing Project (ESP5400 release)54 or 1000 Genome (1000G, 2010_07 release)55 CEU population, were also excluded (“Rare Variant” filter). Finally, we removed any variant that was not shared by all sequenced affected individuals of at least one family, prior to Sanger verification (“Family Segregation” filter).
Post-Sanger Filtering Strategy
Variants were considered verified if Sanger reads had full concordance with NGS reads in all affected individuals and both the forward and reverse reads agreed. Final filtering involved excluding the following: variants outside of the linkage region (“Linked locus” filter); variants with a minor allele frequency >0.1% in the ESP6500 release or 1000G 2012_04 release (“Very Rare Variant” filter); variants not within the exonic sequencing target regions (“Targeted Exome” filter); and variants that did not alter the amino acid sequence of the product protein (“Protein Alteration” filter). The aim of this filtering strategy was to narrow down the list of variants to a list of very rare protein-altering candidate variants within our linkage locus. This was, however, carried out in a step-wise fashion with verification of the excluded variants at each step for potential biological function to ensure we are not excluding any biologically significant variants.
Combined Annotation-Dependent Depletion C-Score
After implementing the above filtering schema, we used scaled C-scores from the Combined Annotation-Dependent Depletion (CADD) method to rank the variants remaining on our final list by potential deleteriousness22. The CADD C-score provides an integration of 63 different annotations (i.e., allelic diversity, functionality, putative pathogenicity and evolutionary conservation) into one score per variant. For example, the C-score integrates scores from PolyPhen38 and SIFT37 for predicted impact of the variant on protein function, GERP56 and PhyloP57 for evolutionary conservation and a number of other annotations for prioritization of causal variants22. A scaled C-score ≥10 indicates variants predicted to be in the top 10% of the most deleterious possible substitutions in the human genome, a score of ≥20 indicates variants predicted to be in the top 1% and a score of ≥30 indicates variants predicted to be in the top 0.1%. More details on the CADD C-score can be found elsewhere22.
Ethics
Written informed consent was obtained from each subject or from his or her guardian. Approval from local institutional review boards was received at each Gliogene participating institution. This study was conducted in accordance with the Declaration of Helsinki.
References
Adamson, C. et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs 18, 1061–83 (2009).
Ohgaki, H. & Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol 109, 93–108 (2005).
Schwartzbaum, J. A., Fisher, J. L., Aldape, K. D. & Wrensch, M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol 2, 494–503; quiz 1 p following 516 (2006).
Ostrom, Q. T. et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 15 Suppl 2ii1–56 (2013).
Ostrom, Q. T. et al. The epidemiology of glioma in adults: a "state of the science" review. Neuro Oncol 16, 896–913 (2014).
Liu, Y. et al. Genetic advances in glioma: susceptibility genes and networks. Curr Opin Genet Dev 20, 239–44 (2010).
Melin, B. & Jenkins, R. Genetics in glioma: lessons learned from genome-wide association studies. Curr Opin Neurol 26, 688–92 (2013).
Rajaraman, P. et al. Genome-wide association study of glioma and meta-analysis. Hum Genet 131, 1877–88 (2012).
Melin, B. et al. Known glioma risk loci are associated with glioma with a family history of brain tumours -- a case-control gene association study. Int J Cancer 132, 2464–8 (2013).
Shete, S. et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet 41, 899–904 (2009).
Sadetzki, S. et al. Description of selected characteristics of familial glioma patients - results from the Gliogene Consortium. Eur J Cancer 49, 1335–45 (2013).
Scheurer, M. E. et al. Familial aggregation of glioma: a pooled analysis. Am J Epidemiol 172, 1099–107 (2010).
Shete, S. et al. Genome-wide high-density SNP linkage search for glioma susceptibility loci: results from the Gliogene Consortium. Cancer Res 71, 7568–75 (2011).
Malmer, B. et al. GLIOGENE an International Consortium to Understand Familial Glioma. Cancer Epidemiol Biomarkers Prev 16, 1730–4 (2007).
Sun, X. et al. A variable age of onset segregation model for linkage analysis, with correction for ascertainment, applied to glioma. Cancer Epidemiol Biomarkers Prev 21, 2242–51 (2012).
Adikes, R. C., Unrath, W. C., Yengo, C. M. & Quintero, O. A. Biochemical and bioinformatic analysis of the myosin-XIX motor domain. Cytoskeleton (Hoboken) 70, 281–95 (2013).
Edgren, H. et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol 12, R6 (2011).
Stout, J. R. et al. Kif18B interacts with EB1 and controls astral microtubule length during mitosis. Mol Biol Cell 22, 3070–80 (2011).
Lee, Y. M. et al. Cell cycle-regulated expression and subcellular localization of a kinesin-8 member human KIF18B. Gene 466, 16–25 (2010).
Rath, O. & Kozielski, F. Kinesins and cancer. Nat Rev Cancer 12, 527–39 (2012).
Itzel, T. S. P., Maass, T., Krupp, M., Marquardt, J. U., Strand, S., Becker, D., Staib, F., Binder, H., Wang, X. W., Thorgeirsson, S. S., Galle, P. R. & Teufel, A. Translating bioinformatics in oncology: Guilty by profiling meta-analysis and identification of KIF18B and CDCA3 as novel driver genes in liver carcinogenesis. Z Gastroenterol 51, 4–26 (2013).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–5 (2014).
Suri, A. et al. Cancer testis antigens: A new paradigm for cancer therapy. Oncoimmunology 1, 1194–1196 (2012).
Yi, F. et al. SPAG9 is overexpressed in human astrocytoma and promotes cell proliferation and invasion. Tumour Biol 34, 2849–55 (2013).
Takaesu, G. et al. Activation of p38alpha/beta MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J Cell Biol 175, 383–8 (2006).
Sinha, A. et al. Down regulation of SPAG9 reduces growth and invasive potential of triple-negative breast cancer cells: possible implications in targeted therapy. J Exp Clin Cancer Res 32, 69 (2013).
Garg, M. et al. Sperm-associated antigen 9 is a biomarker for early cervical carcinoma. Cancer 115, 2671–83 (2009).
Kanojia, D. et al. Sperm associated antigen 9 plays an important role in bladder transitional cell carcinoma. PLoS One 8, e81348 (2013).
Seleit, I., Bakry, O. A., Samaka, R. M. & Malak, M. A. Immunohistochemical Expression of Sperm-Associated Antigen 9 in Nonmelanoma Skin Cancer. Am J Dermatopathol (2014) In press. 10.1097/DAD.0000000000000126.
Garg, M. et al. Sperm-associated antigen 9: a novel diagnostic marker for thyroid cancer. J Clin Endocrinol Metab 94, 4613–8 (2009).
Wang, Y. et al. Clinical significance and biological roles of SPAG9 overexpression in non-small cell lung cancer. Lung Cancer 81, 266–72 (2013).
Xie, C., Fu, L., Liu, N. & Li, Q. Overexpression of SPAG9 correlates with poor prognosis and tumor progression in hepatocellular carcinoma. Tumour Biol 35, 7685–91 (2014).
Li, H. et al. SPAG9 is overexpressed in human prostate cancer and promotes cancer cell proliferation. Tumour Biol 35, 6949–54 (2014).
Baser, E. et al. Sperm-associated antigen 9 is a promising marker for early diagnosis of endometrial cancer. Asian Pac J Cancer Prev 14, 7635–8 (2013).
Forsyth, P. A. et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 79, 1828–35 (1999).
Apodaca, G. et al. Expression of metalloproteinases and metalloproteinase inhibitors by fetal astrocytes and glioma cells. Cancer Res 50, 2322–9 (1990).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–81 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–9 (2010).
Llanos, S., Efeyan, A., Monsech, J., Dominguez, O. & Serrano, M. A high-throughput loss-of-function screening identifies novel p53 regulators. Cell Cycle 5, 1880–5 (2006).
Song, Q. et al. Gene expression analysis on a photodiode array-based bioluminescence analyzer by using sensitivity-improved SRPP. Analyst 135, 1315–9 (2010).
Mutel, E. et al. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol 54, 529–37 (2011).
Lal, A. et al. A public database for gene expression in human cancers. Cancer Res 59, 5403–7 (1999).
Egland, K. A. et al. High expression of a cytokeratin-associated protein in many cancers. Proc Natl Acad Sci U S A 103, 5929–34 (2006).
Hu, L., Crowe, D. L., Rheinwald, J. G., Chambon, P. & Gudas, L. J. Abnormal expression of retinoic acid receptors and keratin 19 by human oral and epidermal squamous cell carcinoma cell lines. Cancer Res 51, 3972–81 (1991).
Shao, M. M. et al. Keratin expression in breast cancers. Virchows Arch 461, 313–22 (2012).
Paunu, N. et al. A novel low-penetrance locus for familial glioma at 15q23-q26.3. Cancer Res 62, 3798–802 (2002).
Robertson, L. B. et al. Survey of familial glioma and role of germline p16INK4A/p14ARF and p53 mutation. Fam Cancer 9, 413–21 (2010).
Reid, J. G. et al. Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 15, 30 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–8 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Futreal, P. A. et al. A census of human cancer genes. Nat Rev Cancer 4, 177–83 (2004).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–20 (2013).
Genomes Project, C. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6, e1001025 (2010).
Cooper, G. M. et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15, 901–13 (2005).
Acknowledgements
This work was supported by grants from the National Institutes of Health and NHGRI, Bethesda, Maryland (R01CA119215, R01CA070917, R01CA52689, P50097257, R01CA126831, U54HG003273, P30CA125123, K23CA158148, 5 R01CA138836, 5 P30 CA125123, R25NS070694). Additional support was provided by the McNair Medical Institute, the Population Sciences Biorespository at Baylor College of Medicine, the American Brain Tumor Association and The National Brain Tumor Society. We would also like to thank the patients and their families for participating in this research.
Author information
Authors and Affiliations
Contributions
A.J. and E.S.A. wrote the main text of the manuscript with significant input from M.N.A., S.N.J., G.N.A., J.S.B.S., C.I.A., D.M.M., B.S.M. and M.L.B. A.J. prepared figures 1-2 and G.N.A. prepared figure 3. A.J., E.S.A., M.N.B., G.N.A., Y.L., S.T., S.E.P., S.S., C.I.A., B.S.M. and M.L.B. contributed to the analysis with substantial input from all other authors. All authors reviewed the manuscript and were involved in study design and conduct.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Targeted Sequencing in Chromosome 17q Full CADD Output
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jalali, A., Amirian, E., Bainbridge, M. et al. Targeted Sequencing in Chromosome 17q Linkage Region Identifies Familial Glioma Candidates in the Gliogene Consortium. Sci Rep 5, 8278 (2015). https://doi.org/10.1038/srep08278
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08278
This article is cited by
-
RUNDC1 inhibits autolysosome formation and survival of zebrafish via clasping ATG14-STX17-SNAP29 complex
Cell Death & Differentiation (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
