
Abstract
Non-alcoholic fatty liver disease (NAFLD) has recently been considered to be under the influence of the gut microbiota, which might exert toxic effects on the human host after intestinal absorption and delivery to the liver via the portal vein. In this study, the composition of the gut microbiota in NAFLD patients and healthy subjects was determined via 16S ribosomal RNA Illumina next-generation sequencing. Among those taxa displaying greater than 0.1% average abundance in all samples, five genera, including Alistipes and Prevotella, were significantly more abundant in the gut microbiota of healthy subjects compared to NAFLD patients. Alternatively, Escherichia, Anaerobacter, Lactobacillus and Streptococcus were increased in the gut microbiota of NAFLD patients compared to healthy subjects. In addition, decreased numbers of CD4+ and CD8+ T lymphocytes and increased levels of TNF-α, IL-6 and IFN-γ were detected in the NAFLD group compared to the healthy group. Furthermore, irregularly arranged microvilli and widened tight junctions were observed in the gut mucosa of the NAFLD patients via transmission electron microscopy. We postulate that aside from dysbiosis of the gut microbiota, gut microbiota-mediated inflammation of the intestinal mucosa and the related impairment in mucosal immune function play an important role in the pathogenesis of NAFLD.
Similar content being viewed by others


Introduction
Recently, non-alcoholic fatty liver disease (NAFLD) has emerged as the most common hepatic disorder in many countries. NAFLD is characterized by a broad spectrum of hepatic pathology that is closely associated with obesity and ranges from simple steatosis (SS) to non-alcoholic steatohepatitis (NASH) and even cirrhosis1. The pathophysiology of NAFLD has been proposed to occur as a result of multiple hits2. Insulin resistance has been identified as a crucial factor in NAFLD. Other factors, including genetic determinants, nutritional factors, adipose tissue and the immune system, may be necessary for the manifestation and progression of NAFLD. Previous studies have provided evidence that the gut microbiota is associated with host metabolism and might play an important role in the pathogenesis of NAFLD3,4.
The human intestinal tract contains approximately 1014 bacteria. This highly diverse and dense gut microbiota plays crucial roles in intestinal health, including the digestion of food, the protection of mucosal surfaces and crosstalk with the immune system of the host. Recent studies have shown that the gut microbiota is involved in the pathogenesis of NAFLD via several mechanisms. First, the gut microbiota are known to regulate metabolism and energy extraction5, which is supported by the finding that treatment of the gut microbiota from high-fat diet-fed mice with a high level of pro-inflammatory cytokines promotes the development of NAFLD6. Second, the gut microbiota share a highly coevolutionary relationship with the immune system7 and impaired immune function has been observed during the progression of NAFLD8. Furthermore, colonic bacteria are a source of many metabolic products, including endogenous ethanol and other endotoxins that may exert toxic effects on the human host after intestinal absorption and delivery of these toxins to the liver via the portal vein, which also induces hepatic steatosis and steatohepatitis9.
Mouzaki et al.10 reported the differences in the gut microbiota of subjects with SS, NASH and healthy controls. They found an inverse association between the presence of NASH and the percentage of Bacteroidetes. Zhu et al.11 showed that NASH was associated with higher levels of ethanol-producing bacteria and serum ethanol. However, some questions remain unclear. What is the precise microbial composition of the gut microbiota in NAFLD patients? Does gut microbiota-mediated intestinal mucosa inflammation impact the NAFLD status? Is the intestinal mucosa barrier impaired in NAFLD patients displaying dysbiosis of the gut microbiota?
To resolve these issues, in this study, we applied Illumina sequencing of 16S rRNA gene V3 region amplicons to profile the overall structure and the composition of the microbiota of fecal samples from 53 NAFLD patients and 32 healthy subjects. Additionally, we evaluated characteristics of CD4+ T and CD8+ T lymphocytes, proinflammatory cytokines (TNF-α, IL-6 and IFN-γ) and intestinal tight junction. Our results revealed striking differences in the fecal microbial population patterns and in the intestinal mucosa barrier between the two groups, suggesting an important role of gut bacteria in liver health. The distinct composition of the gut microbiota between NAFLD patients and healthy subjects could provide a target for intervention or a marker for disease.
Results
Clinical characteristics
A total of 85 participants were recruited between May and October of 2013, including 53 NAFLD patients and 32 healthy subjects. The characteristics and clinical data for these participants are summarized in Table 1. The NAFLD patients displayed a significantly higher BMI (P < 0.01). Alanine transaminase (ALT) and aspartate transaminase (AST) were significantly higher in NAFLD patients than in healthy subjects (P < 0.01). All NAFLD patients displayed sonographic or histologic evidence of hepatic steatosis but not cirrhosis or portal hypertension.
Differences in fecal microbial communities between healthy and NAFLD groups based on taxonomy comparisons.
We analyzed the gut microbiota from the fecal samples of NAFLD patients and healthy subjects based on 16S rRNA gene sequencing. After applying strict trimming criteria to exclude low-quality reads, a total of 51,516,282 reads displaying acceptable quality were obtained, with an average of 606,074 reads per sample. The Shannon index was used to assess the diversity of the fecal microbiota in the NAFLD and healthy groups. No significant difference in the biodiversity of the microbiota was found (Fig. 1).

Estimates of bacterial diversity as assessed by the Shannon index.
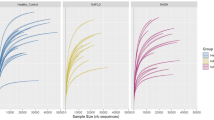
To obtain an overview of the microbial community, we performed a multivariate analysis method, PLS-DA, based on the relative abundance of genera that comprised at least 0.1% of the microbial community, revealing sharp clustering of the microbiome sequence data from all 85 samples (Fig. 2). Most of the samples were clustered by different groups.

The overall structure of the gut microbiota of all samples.
The PLS-DA score plots were based on the relative abundance of the genera displaying greater than 0.1% average abundance. The green circles represent the 53 NAFLD patients and the red circles represent the 32 healthy subjects.
The taxonomic identity of the reads was assigned using the Ribosomal Database Project (RDP) Classifier 2.3. At the phylum level, 20 phyla were detected in all of the samples and as expected, Bacteroidetes and Firmicutes were the predominant phyla (Fig. 3A). The relative abundance of Bacteroidetes was 50.8% and 47.4% in the healthy and NAFLD groups, respectively and the relative abundance of Firmicutes was 41.1% and 42.7%, respectively. No significant differences in the two predominant phyla were observed between the healthy subjects and the NAFLD patients. Another four phyla, Proteobacteria, Actinobacteria, Fusobacteria and Verrucomicrobia, displayed >0.1% abundance in both groups. Of the remaining phyla, only Lentisphaerae was significantly different between the two groups. The healthy group contained a significantly higher abundance of Lentisphaerae than the NAFLD group (P < 0.05).

Comparison of the relative abundance at different taxonomic levels between the 53 NAFLD patients and the 32 healthy subjects.
(A) Average phylum distribution (B) Primary genus composition in the two groups. *P < 0.05 and **P < 0.01.
Within the phylum Firmicutes, the genus Clostridium XI within the family Peptostreptococcaceae displayed a significant increase of approximately 2-fold in the NAFLD group compared to the healthy group (P < 0.01). Moreover, an increase was observed in genus Anaerobacter-related sequences, which belonged to the Clostridiaceae family I in the NAFLD group (P < 0.01) (Fig. 3B). The often pathogenic genus Streptococcus displayed a significant >3-fold increase in the NAFLD group (P < 0.05). Notably, the genus Lactobacillus was also found to be significantly higher in the NAFLD group (P < 0.05). In contrast, Ruminococcaceae was the only family that displayed a higher abundance in the healthy group in this phylum. Two genera in the Ruminococcaceae family, Oscillibacter and Flavonifractor, were increased significantly in the healthy group compared to the NAFLD group (both P < 0.05).
Within the phylum Bacteroidetes, the genus Odoribacter displayed a significantly higher abundance in the healthy group compared to the NAFLD group (P < 0.01), accounting for most of the increased representation of the family Porphyromonadaceae in the healthy group. Another genus, Alistipes, which belongs to the family Rikenellaceae in this phylum, was significantly increased in healthy subjects compared to the NAFLD patients (P < 0.01). There was a 1.6-fold increase in the abundance of the genus Prevotella in the healthy group, but this difference did not reach statistical significance.
The increased abundance of the alcohol-producing bacteria Escherichia within the phylum Proteobacteria was observed in the microbiota from the NAFLD group compared to those from the healthy group (P < 0.05).
Increased intestinal permeability in NAFLD
We examined the tight junctions in the duodenum under a transmission electron microscope (TEM) to establish an index of the intestinal barrier integrity and to evaluate the duodenum microstructure in the two groups (Fig. 4). The duodenum in the healthy group contained intact tight junctions and much more regularly aligned and extensive microvilli. However, widened tight junctions and irregularly arranged microvilli were observed in the NAFLD group. We measured the tight junction gap at the 43,000× magnification and the NAFLD patients displayed a wider tight junction gap than the healthy subjects (P < 0.01) (Fig. 4B), which indicated a loss of intestinal barrier integrity in the NAFLD patients.

Ultrastructural observation of the tight junctions in the duodenal mucosa (transmission electron microscopy, 43,000×).
(A) Mucosa from a NAFLD patient. (B) Mucosa from a healthy subject. (C) The width of the tight junction gap in 26 NAFLD patients and 10 healthy subjects. *P < 0.05 and **P < 0.01.
The occludin proteins are considered to be the structural backbone of intestinal tight junctions. Immunohistochemistry revealed that the expression level of occludin protein was significantly higher in the intestinal mucosa of healthy subjects than that of NAFLD patients (P < 0.01) (Fig. 5). These data indicated that the intestinal mucosal permeability in NAFLD patients was greater than that in healthy subjects.

Immunohistochemical staining for the occludin protein in the duodenal mucosa (light microscopy, 100×).
The proportion of the occludin expression-positive area was scored as follows: 0 (none), 1 (1–25%), 2 (26%–50%), 3 (51%–75%) and 4 (≧76%). The staining intensity was graded according to the following criteria: 0 (no staining), 1 (weak staining, yellow), 2 (moderate staining, brown) and 3 (strong staining, deep brown). (A) Mucosa from a NAFLD patient. (B) Mucosa from a healthy individual. (C) The expression of the occludin protein in the duodenal mucosa in 29 NAFLD patients and 25 healthy subjects. *P < 0.05 and **P < 0.01.
CD4+ and CD8+ T lymphocytes in the mucosa
The intestinal lamina propria is the largest immune area of the entire intestinal mucosal immune system, in which the considerable quantity of T lymphocytes play a critical role in immune surveillance of the epithelium. Among the T lymphocytes in the lamina propria, 65%–85% are CD4+ T lymphocytes, the depletion of CD4+ T lymphocytes in the gastrointestinal tract can lead to immune dysfunction and loss of epithelial integrity and increase the risk for microbial translocation, immune activation and disease progression. While CD8+ T lymphocytes can perform immune responses against the target cells which infected by the pathogen and eliminate them eventually. The number of both CD4+ and CD8+ T lymphocytes in the lamina propria of the duodenal mucosa was fewer in NAFLD patients than in healthy subjects (P < 0.01) (Fig. 6).

Immunohistochemical staining for CD4+ and CD8+ T lymphocytes in the lamina propria of the duodenal mucosa.
(A) CD4+ T lymphocytes in mucosa from a NAFLD patient. (B) CD4+ T lymphocytes in mucosa from a healthy subject. (C) CD8+ T lymphocytes in mucosa from a NAFLD patient. (D) CD8+ T lymphocytes in mucosa from a healthy subject. (E) The number of CD4+ and CD8+ T lymphocytes in the lamina propria of the duodenal mucosa in 29 NAFLD patients and 25 healthy subjects. *P < 0.05 and **P < 0.01.
Intestinal inflammation between the healthy and NAFLD groups
We focused specifically on intestinal inflammation by measuring the mucosal expression of pro-inflammatory cytokines via qPCR. The relative expression of TNF-α (P < 0.05) and IL-6 (P < 0.01) mRNA was significantly higher in the mucosa of NAFLD subjects than that of healthy subjects. There was a trend of an increase in the mRNA expression level of IFN-γ in the NAFLD group compared to the healthy group, but this difference did not reach statistical significance (P = 0.051) (Fig. 7).

Relative mRNA expression levels of TNF-α, IL-6 and IFN-γ in 28 NAFLD patients and 27 healthy subjects. *P < 0.05 and **P < 0.01.
Discussion
To our knowledge, this is the first study to assess the gut microbiota of adults with NAFLD and the intestinal mucosal barrier as an intestine-mediated factor involved in the development of NAFLD. A close association exists between the gut and liver, referred to as the “gut-liver axis”12. This relationship has been based on the evidence that more than 70% of the blood supply to the liver is derived from the portal vein, the direct venous outflow of the intestine. An impaired gut mucosal barrier exposes the liver to gut-derived toxic factors and disrupted liver physiology may induce gut dysfunction13. Aside from the concept that insulin resistance is characterized by complex interactions between genetic determinants, nutritional factors and lifestyle, it is becoming increasingly recognized that there are strong associations between the intestinal microbiota, the mucosal barrier and NAFLD4,14. In this study, we provided additional detail regarding the gut microbiota in NAFLD patients and healthy subjects using Illumina next-generation sequencing technologies. In addition, we explored the CD4+ and CD8+ T lymphocyte profiles, the tight junction protein characteristics and the pro-inflammatory cytokine levels in the intestinal mucosa of a selected group of NAFLD patients and healthy subjects.
We characterized the different patterns of the gut microbiota of NAFLD and healthy subjects. At the phylum level, we identified an increased abundance of Lentisphaerae in the NAFLD group and at the genus level, we observed increases in the abundance of Lactobacillus and Anaerobacter in the NAFLD group. Interestingly, Lactobacillus, which is often used as a probiotic, was also reported to be higher in NAFLD patients by Raman9. To further characterize the increased abundance of Lactobacillus in NAFLD patients, RT-PCR was performed, but no significant differences were detected (P > 0.05). We hypothesize that the metabolic pathway and the subsequent production of this bacterial genus may change the gut microenvironment, which could be involved in the progression of NAFLD.
The involvement of gut microbiota in the development of low-grade inflammation was demonstrated to be associated with obesity and NAFLD15,16,17. Here, we detected increased intestinal permeability and inflammation in NAFLD patients, accompanied by dysbiosis of the gut microbiota. At the genus level, we observed a high abundance of Escherichia in the NAFLD group. Zhu et al.11 reported that members of the genus Escherichia are more abundant in patients with NASH, which was associated with increased blood ethanol concentrations. Endogenous alcohol produced by alcohol-producing bacteria (e.g., Escherichia coli) is known to increase gut permeability. Moreover, Escherichia was reported to be associated with inflammation. Small et al.18 found that persistent infection with adherent-invasive E. coli leads to chronic inflammation. Strains of E. coli that contain the polyketide synthase (pks) genomic island induced increased delivery of pks products, leading to epithelial DNA damage19 and these pathogenic strains were found to be increased in inflammatory bowel disease (IBD) patients20.
Based on these findings, we propose that the high level of Escherichia is an important factor for NAFLD. Another notable finding was that the abundance of inflammation-associated Streptococcus was increased in the NAFLD group. Pathogenic species of Streptococcus, such as Streptococcus bovis and Streptococcus faecalis, have been found to be associated with IBD, which supports their potential role as proinflammatory bacteria21,22. Furthermore, our finding indicated that proinflammatory cytokines, such as TNF-α, IL-6 and IFN-γ, were elevated in the NAFLD gut mucosa biopsies, accompanied by irregularly arranged microvilli and widened tight junctions in the intestinal mucosa. Several lines of evidence further suggest that dysbiosis of the gut microbiota, especially of proinflammatory bacteria, is consistently associated with an elevated level of proinflammatory cytokines and, thus, with gut permeability, which promotes the delivery of bacterial components to the liver during the pathogenesis of NAFLD.
In contrast, two genera of the family Ruminococcaceae displayed significantly higher abundance in the healthy group than in the NAFLD group. This finding is in agreement with previous results in the field of the gut microbiota of nonalcoholic steatohepatitis patients, which demonstrated a lower abundance of the family Ruminococcaceae9,11. The gram-positive Ruminococcaceae, which are commonly found in the intestines of mammals, display the ability to degrade cellulose and hemicellulose in plant material. These compounds are subsequently fermented and converted to short chain fatty acids (SCFAs), which can be absorbed and used for energy by the host23. The protective role of SCFAs against gut inflammation has been well demonstrated24. Furthermore, the genera Alistipes and Prevotella were found to be more abundant in the healthy subjects than in the NAFLD patients. Notably, Alistipes belongs to the family Rikenellaceae, which was also decreased in NAFLD patients based on a recent study11. Alternatively, Prevotella, which uses xylan, xylose and carboxymethylcellulose to produce high levels of SCFAs25, was previously found to be predominant in the fecal microbiota of subjects consuming a long-term low fat/high fiber diet26,27. It has been suggested that these bacterial species may exert protective effects against the development of NAFLD. Interestingly, our results for Prevotella disagreed with those of Zhu et al.11, who showed increased Prevotella abundance in children with obesity or NASH compared to normal subjects. These results may reflect the differences in environmental and dietary factors between these two study cohorts. In addition, because we focused on adults rather than children, age may have also caused the differences in these results.
It is important to assess the intestinal mucosal barrier via mucosa biopsy, as one of the most challenging aspects in determining the inflammation and immune response of mucosa biopsy is understanding the relationship between the gut and the liver. To evaluate the effect of the gut microbiota on immune function, we measured the profiles of CD4+ and CD8+ T lymphocytes. The levels of CD4+ and CD8+ T cells were lower in NAFLD patients than in healthy subjects. Our previous study found that intestinal immune function was dysregulated by a decreased CD4/CD8 ratio in Peyer's patches of HFD mice during the late stage of NAFLD8. Furthermore, Kim et al.28 demonstrated that the CD4+ and CD8+ T cell counts were reduced in obese HFD fed mice. Studies have shown that obesity impairs immune responses, causing lymphopenia in obese humans29. T cell populations and their functions were reduced in human obesity and were related to the elevated TNF-α levels in obese humans30, which is in agreement with our finding. Our findings and these reports further indicated that intestinal immune function is impaired in NAFLD.
We concluded that the impaired intestinal immune function in NAFLD patients may be associated with alterations in the intestinal microbiota and in lipopolysaccharides, the products of dead Gram-negative bacteria in the gut. Lipopolysaccharide has been reported to induce apoptosis in lymphocytes in vivo31,32. In this regard, the loss of lymphocytes from the intestinal mucosa in our study may be associated with the production of metabolic endotoxins due to dysbiosis of the gut microbiota. Therefore, we hypothesize that the dysbiosis of the gut microbiota leads to the intestinal inflammation and the related impaired immune function and that the subsequent increase in intestinal permeability further mediates the pathogenesis of NAFLD via the gut-liver axis.
In conclusion, the crucial role of the gut microbiota in host health is increasingly being recognized, but the involvement of the gut microbiota and the mucosal barrier on the development of NAFLD had yet to be explored in detail. Here, we identified the specific bacterial species associated with the phenotypes of NAFLD and investigated intestinal immune function and the mucosal barrier in NAFLD patients and healthy subjects. Based on the highly detailed data provided by Illumina sequencing, we elucidated the landscape of the gut microbial community, including rare bacterial species, in NAFLD patients and healthy subjects. In addition, we showed that dysbiosis of the gut microbiota and impairment of the intestinal mucosal barrier interferes with the cross-talk between the gut and the liver, which may result in a second hit to NAFLD patients. This study provides new perspectives in the field of the gut-liver axis, as intestinal bacteria have been associated with liver health. Future studies should be performed to determine whether modulating the gut microbiota represents a novel strategy for interrupting NAFLD progression.
Methods
Patients and sample collection
This cross-sectional study was approved by the Conjoint Health Research Ethics Board of Peking University People's Hospital and informed consent forms were signed by all of the subjects prior to participation in this study. All experiments were performed in accordance with the approved guidelines and regulations.
A total of 53 NAFLD patients was recruited at the Peking University People's Hospital. All patients were confirmed to exhibit NAFLD based on evidence of hepatic steatosis via either imaging or histology33. Additionally, 32 healthy subjects were invited to participate as controls. The subjects were excluded if they displayed significant alcohol consumption (the definition of significant alcohol consumption has been inconsistent and ranged from >1 alcoholic beverage (10 grams of alcohol per one drink unit) per day to >40 grams per day). The subjects who exhibited HBV or HCV infection, autoimmune liver disease, hepatic cell carcinoma, or gastrointestinal disease or had consumed antibiotics or probiotics within the previous 3 months were not included. Females who were pregnant or lactating were excluded, as well.
To reduce the effect of diet on the composition of the gut microbiota, general 7-day dietary restrictions were imposed on the participants, including no peppery food and no yogurt intake and appropriate fat intake (fat calorie intake was no more than 35% of the total calories). After providing written informed consent, all subjects were contacted for detailed instructions on how to complete the 7-day dietary restrictions and to collect and transport the stool sample. The stool sample was collected at the end of that week. The stool samples were frozen immediately after collection and were stored at −80°C until DNA extraction.
Among the 85 subjects, 65 volunteers, including 35 NAFLD patients and 30 healthy subjects, agreed to undergo gastroscopy for physical examination and signed their appropriate informed consent. In all cases, the stool samples were collected prior to gastroscopy. The biopsy samples were obtained during gastroscopy for transmission electron microscopy, immunohistochemistry and RT-PCR assays. On the day prior to gastroscopic procedure, a blood sample was collected to measure the metabolic and hepatic parameters.
Illumina library generation
DNA was extracted from stool samples using the QIAamp DNA Stool Mini kit protocol (Qiagen, German). The V3 region of the 16S rRNA gene was amplified using 341F (5′-CCTACGGGAGGCAGCAG-3′) and 534R (5′-ATTACCGCGGCTGCTGG-3′). The V3-specific primer regions were associated with the adaptor and the sequences, which were complementary to the Illumina forward and reverse sequencing primers (within the reverse primer containing a 6-bp barcode, allowing for sample sorting) (Supplementary Fig. S1). Each PCR product of the appropriate size was purified and quantified using a Qubit fluorometer and then added to a master pool of DNA for 125-nucleotide paired-end read assembly using the Hiseq 2000 genome analyzer (Illumina Hiseq 2000, USA). The 96 libraries accounted for approximately 70% of the total DNA sent for sequencing in the two lanes; other genomic samples unrelated to this study accounted for approximately 30% of the total DNA as a control.
Bioinformatics
The Illumina reads were sorted into different samples according to their index sequence. The raw sequence reads were trimmed according to the following criteria: (1) removal of the primers based on complete matching to the raw read, (2) trimming of the reads until 5 continuous bases were greater than Q20, (3) a length of at least 50 nt, (4) exclusion of the reads mapped to the human genome using SOAP software, (5) assembly of the overlapping paired-end reads to generate assembled contigs. If non-assembled paired-end reads were discovered, these reads were concatenated to 8 “N”s between read 1 and the reverse complement of read 2.
The RDP Classifier was used to assign all of the 16S rRNA gene sequences to a taxonomical hierarchy using a confidence threshold of 50%. Both the assembled and non-assembled reads were analyzed. The relative abundances of the various phyla, families and genera in each sample were computed and compared between the NAFLD patients and the healthy subjects. The comparison of the bacterial diversity of these samples was performed using the Shannon index. The reads displaying greater than 0.1% abundance in both groups were further analyzed via partial least-squares discriminant analysis (PLS-DA) to visualize the difference between two groups using the standard Simca-p+ software (version 12.0; http://www.umetrics.com/).
Assessment via transmission electron microscopy
A total of 26 NAFLD and 10 healthy biopsy samples was subjected to transmission electron microscopy. The biopsy samples were immediately fixed using 3% glutaraldehyde and kept at 4°C for 2 hours, then post-fixed with osmium tetroxide and dehydrated with gradient alcohol and finally the tissues were infiltrated with a 1:1 solution of Epon 812 and epoxypropane, then embedded in Epon 812 resin. Ultrathin sections were stained with uranyl acetate and lead citrate and were photographed using a transmission electron microscopy (Philips Tecnai-12 Biotwin, Netherlands).
High-magnification (43,000×) images were captured to evaluate the ultrastructure of the tight junctions in the duodenum. We captured each image under the same brightness and contrast settings. The width of the tight junctions was blindly measured and averaged by two examiners according to a standard protocol34.
Immunohistochemistry
Biopsy samples were obtained from 29 NAFLD patients and 25 healthy subjects for immunohistochemistry. Immunohistochemistry was performed on serial 3-mm sections of the formalin-fixed paraffin-embedded duodenum biopsy. The slides were treated with mouse anti-CD4 (dilution 1:50) or rabbit anti-CD8 (dilution 1:50) overnight at 4°C, or rabbit anti-occludin antibody (dilution 1:80) for 2 h at room temperature, finally visualized with diaminobenzidine, counterstained with hematoxylin, dehydrated and mounted. The brown staining of CD4 and CD8 on the cell membrane and of occludin on the cell membrane or cytoplasm was classified as positive staining. The negative controls were handled in the same manner but were incubated in PBS instead of the primary antibody. External positive controls were always included in the batch of slides.
The immunostaining was reviewed and scored independently by two histopathologists. We counted the average number of CD4+or CD8+ T lymphocytes in the lamina propria in 3 representative high-power fields (HPFs) and the results were presented as the average number of cells/HPF. The staining index for the occludin protein was calculated as the product of the immunopositive area and the staining intensity score.
RT-PCR
A total of 28 NAFLD and 27 healthy duodenum biopsy samples was subjected to RT-PCR to assess the mRNA expression of inflammatory cytokines, including IL-6, TNF-α and IFN-γ. The primer sequences are summarized in Supplementary Table S1. The expression of each inflammatory cytokine was assessed relative to the housekeeping gene GAPDH. Each sample was evaluated in duplicate. The results were normalized to the expression of the GAPDH gene.
Fecal DNA from 53 NAFLD and 32 healthy samples were subjected to RT-PCR assays to determine the abundance of the genus Lactobacillus based on the detection of 16S rRNA genes. The primer sequences were as follows: lac-F (5′-GCAGCA GTAG GGAATCTTCC ACAAT-3′) and lac-R (5′-GCTCGCTTTA CGCCCAAT-3′). The copy number of the target DNA was determined via comparison to the plasmid DNA dilution series standard curves. Bacterial quantity was expressed as log10 bacteria per gram of stool.
Statistical analysis
The data are presented as the mean ± s.d. Differences in the gut microbiota at different levels between the NAFLD and healthy groups were analyzed using Student's t-test. The Mann-Whitney U-test was performed to determine the changes in the numbers of CD4+ and CD8+ T lymphocytes, the level of occludin protein and the relative expression levels of TNF-α, IL-6 and IFN-γ. Values of P < 0.05 were considered to be statistically significant.
Accession number
The sequence data in this study have been deposited in the GenBank Sequence Read Archive under accession number SRP041721.
References
Torres, D. M., Williams, C. D. & Harrison, S. A. Features, diagnosis and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 10, 837–858 (2012).
Tilg, H. & Moschen, A. R. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846 (2010).
Abu-Shanab, A. & Quigley, E. M. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7, 691–701 (2010).
Moschen, A. R., Kaser, S. & Tilg, H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab 24, 537–545 (2013).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Le Roy, T. et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794 (2013).
Hooper, L. V., Littman, D. R. & Macpherson, A. J. Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012).
Su, L. et al. Intestinal immune barrier integrity in rats with nonalcoholic hepatic steatosis and steatohepatitis. Chin Med J (Engl) 125, 306–311 (2012).
Raman, M. et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 11, 868–875 e861–863 (2013).
Mouzaki, M. et al. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 58, 120–127 (2013).
Zhu, L. et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609 (2013).
Son, G., Kremer, M. & Hines, I. N. Contribution of gut bacteria to liver pathobiology. Gastroenterol Res Pract 2010, 1–13 (2010).
Compare, D. et al. Gut--liver axis: the impact of gut microbiota on non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 22, 471–476 (2012).
Miele, L. et al. Gut-liver axis and microbiota in NAFLD: insight pathophysiology for novel therapeutic target. Curr Pharm Des 19, 5314–5324 (2013).
Fallucca, F., Porrata, C., Fallucca, S. & Pianesi, M. Influence of diet on gut microbiota, inflammation and type 2 diabetes mellitus. First experience with macrobiotic Ma-Pi 2 diet. Diabetes Metab Res Rev 30 Suppl 1, 48–54 (2014).
Cani, P. D. et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 58, 1091–1103 (2009).
Cani, P. D., Osto, M., Geurts, L. & Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 3, 279–288 (2012).
Small, C. L., Reid-Yu, S. A., McPhee, J. B. & Coombes, B. K. Persistent infection with Crohn's disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat Commun 4, 1957 (2013).
Cuevas-Ramos, G. et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci U S A 107, 11537–11542 (2010).
Prorok-Hamon, M. et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 63, 761–770 (2014).
Al-Jashamy, K., Murad, A., Zeehaida, M., Rohaini, M. & Hasnan, J. Prevalence of colorectal cancer associated with Streptococcus bovis among inflammatory bowel and chronic gastrointestinal tract disease patients. Asian Pac J Cancer Prev 11, 1765–1768 (2010).
Cucchiara, S., Iebba, V., Conte, M. P. & Schippa, S. The microbiota in inflammatory bowel disease in different age groups. Dig Dis 27, 252–258 (2009).
Scheppach, W. & Weiler, F. The butyrate story: old wine in new bottles? Curr Opin Clin Nutr Metab Care 7, 563–567 (2004).
Kles, K. A. & Chang, E. B. Short-chain fatty acids impact on intestinal adaptation, inflammation, carcinoma and failure. Gastroenterology 130, S100–105 (2006).
Flint, H. J., Bayer, E. A., Rincon, M. T., Lamed, R. & White, B. A. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol 6, 121–131 (2008).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108 (2011).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107, 14691–14696 (2010).
Kim, C. S. et al. Visceral fat accumulation induced by a high-fat diet causes the atrophy of mesenteric lymph nodes in obese mice. Obesity (Silver Spring) 16, 1261–1269 (2008).
Tanaka, S. et al. Impaired immunity in obesity: suppressed but reversible lymphocyte responsiveness. Int J Obes Relat Metab Disord 17, 631–636 (1993).
Tanaka, S., Isoda, F., Ishihara, Y., Kimura, M. & Yamakawa, T. T lymphopaenia in relation to body mass index and TNF-alpha in human obesity: adequate weight reduction can be corrective. Clin Endocrinol (Oxf) 54, 347–354 (2001).
Norimatsu, M. et al. Lipopolysaccharide-induced apoptosis in swine lymphocytes in vivo. Infect Immun 63, 1122–1126 (1995).
Nielsen, J. S. et al. Rough-Form-Lipopolysaccharide Increase Apoptosis in Human CD4(+) and CD8(+) T-Lymphocytes. Scand J Immunol 75, 193–202 (2012).
Chalasani, N. et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases and American College of Gastroenterology. Gastroenterology 142, 1592–1609 (2012).
Cong, X. et al. Activation of transient receptor potential vanilloid subtype 1 increases expression and permeability of tight junction in normal and hyposecretory submandibular gland. Lab Invest 92, 753–768 (2012).
Acknowledgements
This work was supported by funding from The Capital Health Research and Development Special Fund (grant no. 2011-4022-06).
Author information
Authors and Affiliations
Contributions
The project was conceived and designed by Y.L. W.J., N.W., X.W. and Y.C. contributed equally to this work. N.W. performed the DNA extraction and the microbial data analysis; W.J. and X.W. collected the clinical samples and performed the immunohistochemistry, transmission electron microscopy and RT-PCR assays together; Y.C. collected the clinical samples and performed the RT-PCR assay. Y.Z., X.Q. and Y.H. helped to collect the clinical samples; Y.Z. and X.Q. helped to perform and analyze the laboratory experiments; J.L. analyzed the results; and N.W. and Y.L. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Jiang, W., Wu, N., Wang, X. et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep 5, 8096 (2015). https://doi.org/10.1038/srep08096
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08096
This article is cited by
-
Exploring the interplay of gut microbiota, inflammation, and LDL-cholesterol: a multiomics Mendelian randomization analysis of their causal relationship in acute pancreatitis and non-alcoholic fatty liver disease
Journal of Translational Medicine (2024)
-
Immunology of gut microbiome and liver in non-alcoholic fatty liver disease (NAFLD): mechanisms, bacteria, and novel therapeutic targets
Archives of Microbiology (2024)
-
Non-alcoholic fatty liver disease in patients with morbid obesity: the gut microbiota axis as a potential pathophysiology mechanism
Journal of Gastroenterology (2024)
-
Comparative efficacy of oral drugs for chronic radiation proctitis — a systematic review
Systematic Reviews (2023)
-
Impaired flux of bile acids from the liver to the gut reveals microbiome-immune interactions associated with liver damage
npj Biofilms and Microbiomes (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
