
Abstract
Tyrosinase is involved in melanin biosynthesis and the abnormal accumulation of melanin pigments leading to hyperpigmentation disorders that can be treated with depigmenting agents. A natural product T1, bis(4-hydroxybenzyl)sulfide, isolated from the Chinese herbal plant, Gastrodia elata, is a strong competitive inhibitor against mushroom tyrosinase (IC50 = 0.53 μM, Ki = 58 ± 6 nM), outperforms than kojic acid. The cell viability and melanin quantification assay demonstrate that 50 μM of T1 apparently attenuates 20% melanin content of human normal melanocytes without significant cell toxicity. Moreover, the zebrafish in vivo assay reveals that T1 effectively reduces melanogenesis with no adverse side effects. The acute oral toxicity study evidently confirms that T1 molecule is free of discernable cytotoxicity in mice. Furthermore, the molecular modeling demonstrates that the sulfur atom of T1 coordinating with the copper ions in the active site of tyrosinase is essential for mushroom tyrosinase inhibition and the ability of diminishing the human melanin synthesis. These results evident that T1 isolated from Gastrodia elata is a promising candidate in developing pharmacological and cosmetic agents of great potency in skin-whitening.
Similar content being viewed by others
Introduction
Melanin is the major cellular component commonly observed in bacteria, fungi, plants and animals responsible for skin color1. It presents as a complex, heterogeneous polyphenol-like biopolymer structure and colors vary from yellow to black2, secreted by melanocyte cells in the basal layer of the dermis3. Normal melanin pigmentation is able to shield from UV radiation, inhibit photocarcinogenesis and affect the synthesis of vitamin D34. In contrast, the abnormal pigmentation, such as senile lentigines, freckles, melasma and other forms of melanin hyperpigmentation, causes serious esthetic problems5,6. The oxidative reactions of the tyrosine catalyzed by tyrosinase mainly contributes to the melanin biosynthesis7.
As a binuclear copper enzyme, tyrosinase (monophenol monooxygenase EC 1.14.14.1) catalyzes two distinct reactions of melanin biosynthesis. It catalyzes phenols to catechols and further oxidizes catechols to quinones8. The tyrosinase contains two copper ions, coordinating with histidine residues in the active site. The two copper ions are critical for the catalytic activities of this enzyme9 and exist in different tyrosinases regardless of their source10,11. Since tyrosinase-catalyzed reaction is highly associated with local hyperpigmentaiton such as ephelide, melasma and lentigo5, discovering of tyrosinase inhibitors are of great importance in cosmetic and medicinal products for the prevention of pigmentation disorders12. Recently, significant efforts have been made to search for the tyrosinase inhibitors with copper chelator ability as whitening and anti-hyperpigment agents13,14,15,16,17,18 and several tyrosinase inhibitors have also been used as depigmentation ingredients of medical products19,20,21,22.
Many tyrosinase inhibitors, such as hydroquinone23,24,25,26, kojic acid20, azelaic acid27,28, electron-rich phenols29 and arbutin have been tested in pharmaceuticals and cosmetics for their capability of preventing overproduction of melanin30,31. Meanwhile, their structure-activity relationship (SAR) analysis have been widely discussed32 (Supplementary Table S1). Hydroquinone is one of the most frequently prescribed ingredients among the conventional skin-whitening agents. However, hydroquinone causes skin irritation33 and it is thought to be mutagenic to mammalian cells34 and cytotoxic to melanocytes. This leads to the use of kojic acid and arbutin as alternative agents, but these agents show poor efficacy in vivo. In addition, due to their adverse side effects, low formulation stability and poor skin penetration35, their use is still limited. The Japanese officials also spurred to ban the use of kojic acid in skin treatment due to its carcinogenicity36. Thus, it is in great need of developing new tyrosinase inhibitors from different sources. In this regard, the natural products have been extensively utilized in the cosmetics industry37,38,39 because of lower adverse side effects and high safety.
Recently, the extracts and isolated compounds of a number of natural sources in particular botanical sources were well characterized with anti-tyrosinase activities40,41,42,43,44,45,46,47,48,49,50,51 and have been accepted as popular skin lightening agents52,53,54,55,56. In this study, we have screened 78 different medicine herbal plants and identified that rhizome of Gastrodia elata is of great potency for tyrosinase inhibition. The rhizome of Gastrodia elata has been applied in for the treatment of dizziness, headaches, vertigo and convulsive illnesses57. The studies of how Gastrodia elata prevents the neuronal damage have been performed as well58,59,60,61,62,63, but its efficacy on tyrosinase inhibition and melanin biosynthesis has not been thoroughly investigated. Thus, here we aim to isolate the functional components from the rhizome of Gastrodia elata and investigate its inhibitory effect on the mushroom tyrosinase and human melanogenesis. The natural compounds, T1 and T2 extracted from Gastrodia elata and their derivatives T3–T5, exert profound mushroom tyrosinase inhibitory abilities. The bioactive natural product T1, bis(4-hydroxybenzyl)sulfide, with the most striking inhibitory potency against tyrosinase, was chosen as the target compound to characterize its biological effects in tyrosinase inhibition, cell viability, in vitro and in vivo melanin biosynthesis and acute oral toxicity in mice.
Methods
Mushroom tyrosinase inhibition assay and IC50 determination
Tyrosinase inhibition activity was evaluated by using L-tyrosine as the substrate in vitro according to the previous method64. T1 and T2 chemicals were obtained from our previous neuroprotective research57. T3, T4 and T5 chemicals were purchased from ACROS Organics (Geel, Belgium). T1 and its analogous compounds (T2–T5) were prepared (dissolved in 1% DMSO) into distinct concentration inhibitor solutions. Briefly, 80 μl of 67 mM potassium phosphate buffer (NaH2PO4-Na2HPO4, pH6.8), 25 μl of desired concentration of inhibitor solution and 125 μl of 5 mM L-tyrosine were mixed and added into each well of a 96-well Elisa plate, incubated at 25°C for 5 minutes. After that 20 μl of 1250 U/ml mushroom tyrosinase solution was added into each well to a final volume 250 μl and incubated at 25°C for another 5 minutes. Furthermore, the amount of dopachrome produced was determined against blank by a spectrophotometer (Varian cary-50 Bio UV-Visible spectrophotometer) at 475 nm for 10 minutes. We recorded dopachrome accumulation in each 10 seconds for 10 minutes. In addition, kojic acid and β-arbutin were used as the positive control at the same concentrations and conditions to those of the tested inhibitors. The reaction correlating with the amount of dopachrome produced was determined by the previously described method64. The tyrosinase activity is calculated with the following equation:

where S denotes the OD475 absorbance of test compound, B is OD475 absorbance of the blank and C represents the OD475 absorbance of control. The dose-dependent inhibition experiments were performed in triplicate to determine the IC50 of the test compounds.
Cell viability and Melanin quantification assay
Normal human epidermal melanocytes (Cascade BiologicsTM (Portland, OR)) were cultured in HMGS (Cascade Biologics) supplemented Medium 254. For experiments, confluent cells were trypsinized and suspended in Melanocyte Growth Medium M2 at 2 × 105 cells/ml. Then the cells were placed in 96-well plates (2 × 104 cells/well) for cell viability assay and 24-well plates (1 × 105/well) for melanin content assay. After 24 hours, the cells were respectively treated with α-arbutin (50 μM), β-arbutin (50 μM), kojic acid (50 μM) and T1 (5 μM and 50 μM) at 37°C and incubated for 72 hours. Cell viabilities were determined by MTT method15,65,66,67. From measurement of melanin contents, 1N hot NaOH (70°C) was used to and dissolved the melanocyte pellets for 1 hour and further centrifuged at 10,000 × g for 10 min. The microplate reader (Molecular Devices Spectra Max M2) was employed to determine the optical density (OD405) of each supernatant.
Zebrafish in vivo assay
Zebrafish in vivo assay was performed according to the previous method68. The collected synchronized zebrafish embryos were arrayed by pipette into a 96-well plate, three embryos per well with 200 μl embryo medium. The prepared inhibitor solutions (in 1% DMSO) were added to the embryo medium from 9 to 57 hpf (hours post fertilization, total 48 h exposure). The positive controls were 0.1, 1 and 10 mM arbutin and 10, 25, 40 and 50 μM kojic acid. Stereomicroscope was employed for observing the effects on the pigmentation of zebrafish. Phenotype-based evaluation of the body pigmentation was performed at 57 hpf. Embryos were deteriorated by forceps, anesthetized in tricaine methanesulfonate solution (Sigma-Aldrich), mounted in 1% methyl cellulose on a depression slide (Aquatic Eco-Systems, Apopka, FL, USA) and photographed under the stereomicroscope Z16 (Leica Microsystems, Ernst-Leitz-Strasse, Germany) for observation. Images capturing and pixel measurement analysis were carried out by our same method described previously69. All the experimental protocols were reviewed and approved by National Sun Yat-sen University Animal Care and Use Committee (Approval 10117). We followed the Guiding Principles in the Care and Use of Animals of the American Physiology Society during zebrafish maintenance and experimentation.
Acute oral toxicity study in mice
An acute oral toxicity study was performed to evaluate the toxicity of T1 in ICR male mice (Strain: Cr1: CD1 (IICR); Source: BioLASCO Taiwan C0., Ltd, Taipei, Taiwan) via single oral administration. 24 mice were used and randomized into 4 groups, each consisting of six males. The treated mice were administered with T1 at dose levels of 1500, 3000 and 6000 mg/kg and the control mice were administered with vehicle control (4% Tween 80 in water for injection (Brand: SIGMA, Lot No.: 44H01211) was used for vehicle control and dosing solution preparation). The dosing volume of 20 ml/kg was adjusted according to the individual body weight recorded prior to dosing. The mice were observed for clinical signs and mortality for 14 days at 0, 1, 2, 3 and 4 hours after dosing. The observations for the mortality (twice daily, at least six hours apart) and clinical signs (once a day) were conducted from SD3 to SD14. All clinical signs were recorded. Body weight records were made on all animals before the start of dosing (SD1), after seven days interval (SD8) and on the final day of the study (SD15). All surviving mice were subjected to gross necropsy at the end of study (SD15). The surviving mice were further exsanguinated and necropsied in the thoracic and abdominal cavities for clinical check under euthanized condition. All experiments were approved by the Animal Care and the Use Committees of Center of Toxicology and Preclinical Sciences, the Development Center for Biotechnology (Approval FR-AC00266E).
Molecular Modeling
Models of the compound T1, T2, T3 and T5 in complex with tyrosinase were generated through molecular docking analysis. In order to predict the positions of T1, T2, T3 and T5 in the active site, we implemented a docking program (GOLD Genetic Optimization for Ligand Docking) (Cambridge Crystallographic Data Center (CCDC), version 5.2) with the Goldscore scoring function. Before docking, the cofactors and all water molecules were removed. The 3D structure of the compound T1, T2, T3 and T5 were generated and optimized by energy minimization using Discovery Studio v.4.0 (Accelrys Software Inc., USA). GOLD was used to dock T1, T2, T3 and T5 into the active site of the X-ray (PDB ID: 2Y9X) and a human tyrosinase model (built by Multiple sequences alignment and Homology modeling of Discovery Studio v.4.0 using the structure of mushroom tyrosinase as a template (PDB ID: 2Y9X) (Supplementary Fig. S1)) with the flexible docking option turned on. To comprehensively examine the docking conformational space, the search efficiency was set at 100%. All other parameters were set as default. Finally, from the 100 docking conformations, the top one with the highest GOLD fitness score was chosen to explore the binding mode of docked compound in the mushroom tyrosinase active site using Goldscore within the GOLD program. The molecular models of the docked compound were displayed using the PyMOL software. (http://www.pymol.org).
Results
Tyrosinase inhibitory potency of T1 and its derivative compounds
Mushroom tyrosinase has been extensively applied for the estimation and characterization of potential tyrosinase inhibitors70. Our group extracted 400 natural compounds from 78 known Chinese herbal plants and the preliminary tyrosinase inhibition test showed that second natural components, T1 and T2, isolated from Gastrodia elata exhibit apparent tyrosinase inhibitory ability. T2, T3, T4 and T5 are T1 derivative compounds. Therefore, we further quantified their inhibitory capacity in IC50 for comparison with those of the reference inhibitors, β-arbutin and kojic acid (Fig. 1). The result shows that the natural compound T2 exhibits equivalent inhibitory ability to that of β-arbutin and T4 shows less potent tyrosinase inhibitory potency (IC50 > 2400 μM). On the other hand, the compounds T3 and T5 display stronger tyrosinase inhibitory abilities than β-arbutin and the natural compound T1 (IC50 = 0.53 μM) exhibits the greatest inhibitory potency among all, even superior to kojic acid (IC50 = 40.69 μM). Moreover, a Dixon plot is used to determine the enzyme-inhibitor inhibition constant (Ki). The Ki value for T1 against mushroom tyrosinase is 58 ± 6 nM (Supplementary Fig. S2), indicating that T1 is a strong competitive inhibitor and is comparable with those have been reported tyrosinase inhibitors71.

Comparison of mushroom tyrosinase inhibitory abilities (IC50) among kojic acid, arbutin, natural product T1 and its derivative T2–T5.
(a) The IC50 values of β-arbutin, kojic acid, natural product T1 and its derivative T2–T5. (b) 2D chemical structure of natural product T1 and its derivative T2–T5.
Cell viability and Melanin quantification assay
The inhibitory effect of T1 on human melanin formation was investigated. The change of melanin content in the normal human melanocyte cells treated with 5 μM and 50 μM of T1 was examined individually to determine the depigmentation activity. The result shows that only 50 μM of T1 sufficiently attenuate 20% melanin content (Fig. 2b), whereas there is little or no decrease of the melanin content at the presence of 50 μM of α- and β-arbutin and kojic. Moreover, to evaluate the cytotoxic effect of T1, MTT assay was applied to investigating whether the T1 compound would induce cell death adversely. The effect of T1 on cell viability was shown in Fig. 2a. At a dose of 5 and 50 μM of T1, cell viability is 99.2% and 97.2% respectively, almost identical to those of α- and β-arbutin and kojic. Thus, it is believed that T1 is non-cytotoxic to normal human melanocyte cells in the concentration ranging from 0 to 50 μM.

The effects of arbutin, kojic acid and T1 on cell viability and melanogenesis of human melanocyte.
(a) The cell viability under the individual treatment of arbutin, kojic acid and T1 determined with 5 μM and 50 μM by MTT assay. (b) The melanin contents of arbutin, kojic acid and T1 treated human melanocytes with 50 μM. DMSO indicates buffer control. ***P < 0.001 versus DMSO control.
Zebrafish (in vivo assay)
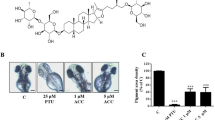
In addition to estimating the effects of T1 on cell viability and human melanogenesis, we further evaluate its anti-pigmentation ability through in vivo. Zebrafish is an particularly useful vertebrate model organism because it possesses similar gene sequences and organ systems to human beings72. With these advantages, the zebrafish system was employed to investigate the in vivo melanogenic inhibition68 and the compound toxicity was also determined simultaneously. The inhibition effects of T1 on the pigmentation of zebrafish were evaluated. Arbutin and kojic acid were used as control groups. With the treatment of 50 μM T1, the pigmentation level of zebrafish obviously decreases about 41.4% (Fig. 3b). However, the de-pigmenting effects at the presence of 0.1, 1 and 10 mM of β-arbutin are individually much lower than that of 50 μM T1. Additionally, T1 also shows a better de-pigmenting effect than kojic acid—at 50 μM, kojic acid decreases 12.9% pigmentation level, while T1 can decrease 3-fold of that (41.4%), as shown in Fig. 3b.

Estimation and comparison of depigmenting effects of β-arbutin, kojic acid and T1 by in vivo zebrafish assay.
(a) Illustration of the pigmentation levels of zebrafish treated with T1 (50 μM) and β-arbutin (0.1 mM, 1 mM and 10 mM). (b) Comparison of the depigmenting effects of kojic acid and T1 at 10, 25, 40 and 50 μM. KA represents Kojic acid.
Acute oral toxicity assay in mice
An acute oral toxicity study was performed to examine the toxicity of T1 in ICR male mice via single oral administration. 24 mice were used and randomized into 4 groups, each consisting of 6 mice. There was no death and abnormal clinical symptom observed in any subject, which lived up to 14 days after the administrations of the T1 compound at the dosage levels of 1500, 3000 and 6000 mg/kg body weight (Fig. 4). The mean body weight and mean body weight gain in mice were calculated, as presented in Fig. 1. The mean body weight growth curves are shown in Fig. 5. There is no difference in body weight as well as body gain observed among T1-treated and control mice and no observable gross lesion was found.

The mean body weight growth curves of T1-treated mice in the acute oral toxicity study.
Body weight of all mice treated with T1 was recorded at the start of dosing (SD 1), weekly interval (SD 8) and the end of the study period (SD 15).

Molecular docking analyses of T1 and T2 toward mushroom tyrosinase.
(a) The docking model of T1 molecule in the active site of mushroom tyrosinase. T1 molecule is colored in cyan and residues interacting with T1 are colored in white and orange (6 histidine residues). (b) The docked T2 molecule (colored in green) interacts with the active site residues (colored in white and orange) of mushroom tyrosinase. (c) 2D schematic representation of the interactions between T1 and active site residues of mushroom tyrosinase. (d) 2D schematic representation of the interactions between T2 and active site residues of mushroom tyrosinase. (The green dash lines in a and b panels donate the hydrogen bond interactions, the yellow ones represent the hydrophobic interactions and the red spheres represent two cooper ions. In 2D schemes, green dash line denotes hydrogen bond interaction; circles in distinct colors represent the relative hydrophobic and/or π-π interactions).
Molecular docking analysis
GOLD molecular docking analysis was applied to investigate the binding interactions of T1, T2, T3 and T5 molecules, respectively with mushroom tyrosinase to elucidate the possible molecular mechanism. In our docking model, the sulfur atom of T1 makes close contacts with the copper ions of tyrosinase (Fig. 5a and c). In addition, two hydrogen bonds were observed between the hydroxyl groups of T1 and Asn260 and His224, respectively. The side chains of residues Glu256, Phe90, Val238 and Phe264 show tightly hydrophobic contacts and/or π-π interactions with the T1 molecule. The molecular docking results of T2, T3 and T5 will be compared and discussed in discussion.
Discussion
The inhibition of tyrosinase, decreasing of melanocyte metabolism and avoiding of UV exposure are ways to reduce melanin synthesis. Among these, tyrosinase inhibitors are of great treatment for pigmentation and developed as cosmetically skin-whitening agents22,31,70,73. In this study, for the purpose of developing a safe and effective skin-whitening agent, we screened about 400 natural products from 78 different of Chinese herbal plants for compounds with inhibitory activity against mushroom tyrosinase. The natural compound T1 isolated from Gastrodia elata shows outstanding inhibitory potency against tyrosinase (IC50 = 0.53 μM, Ki = 58 ± 6 nM), which is more effective than β-arbutin, kojic acid, short peptides74,75 and other known natural compounds (Supplementary Table S1). Our docking model demonstrates that the sulfur atom of T1 makes close contacts with the copper ions in the active site of tyrosinase (Fig. 5a and c), implying that T1 may function as a copper chelator to abolish the tyrosinase activity and this can be evident by our inhibition assay result–T1 is a strong competitive inhibitor of mushroom tyrosinase (Supplementary Fig. S2). However, the natural product T2, with a replacement of the sulfur atom of T1 by an oxygen atom, shows 713-fold decrease in the inhibitory ability (IC50 = 378.11 μM). The docking model shows that T2 applies distinct orientation to interact with the active site residues of mushroom tyrosinase–its hydroxyl group of one benzene ring but not the oxygen atom makes close contacts with one copper ion (Fig. 5b and d). This indicates that the sulfur atom of T1 is of great potential in interacting with copper ions than the oxygen atom of T2 and significant contributes in mushroom tyrosinase inhibition. On the other hand, shortening of the carbon linker which connects the sulfur atom to benzene rings results in the moderate tyrosinase inhibition of T3 (IC50 = 102.35 μM), demonstrating the significance of the degree of freedom of the sulfur atom and benzene rings in tyrosinase inhibition (Supplementary Fig. S3a). In addition, the removal of the hydroxyl groups of T1 (corresponding to T4 molecule) leads to poor tyrosinase inhibition (IC50 > 2400 μM), indicating that the two hydroxyl groups which contribute to the hydrogen bond interactions with Asn260 and His224 (Fig. 5a and c) make extensive contributions to the inhibition ability. Finally, T5, analogous to T1 where hydroxyl groups are substituted by methoxy groups, results in an IC50 = 40.02 μM, suggesting that hydrogen bonds (provided by the hydroxyl groups) are more favorable than the hydrophobic interactions (supported by the methoxy groups) in mushroom tyrosinase inhibition (Supplementary Fig. S3b).
For the purpose of finding an effective and safe whiting substance which can be applied for preventing human hyperpigmentation, the natural product T1 is of great candidacy for evaluating its cell viability and inhibition of melanogenesis. Treatments (5 and 50 μM) of T1 do not show significant cytotoxicity to human normal melanocytes, making it superior to arbutin and kojic acid (Fig. 2). In the human melanocyte system, we also found that melanin production levels apparently decrease by about 20% when treated with 50 μM of T1; however, there is very few or no melanin content decrease observed in the treatment with 50 μM α- and β-arbutin and kojic acid. These results demonstrate that the natural compound T1, taken from the herbal medicine source (Gastrodia elata), apparently attenuates melanogenesis without significant cytotoxicity. This result indicates that the reduced melanin production is not attributable to the cytotoxicity of T1 and T1 can be a safe skin-lightening agent without influencing melanocyte growth. In addition, the molecular docking analysis shows that T1 adapts the same orientation as interacting with mushroom tyrosinase to interact with human tyrosinase (Supplementary Fig. S4). However, the mainly difference is that the hydrophobic and hydrogen-bonding interactions contributed by the active site residues, H244, E256 and F264 in mushroom tyrosinase are not observed in the corresponding active site residues, P505, S515 and I523 of human tyrosinase (Supplementary Fig. S1). This indicates that T1 may be more specific to mushroom instead of human tyrosinase, giving an explanation of T1 significantly inhibits the activity of mushroom tyrosinase but is not highly efficient on reducing human melanin production.
The melanin content of T1-treated zebrafish was investigated. The substantially reduces of skin melanin content in the developed larvae are observed in the T1 treated (between 9–57 hpf, total 48 h exposure) embryos. At the same concentration (50 μM), T1 causes a higher degree of transparency (reducing around 41.4% of pigmentation production) than those seen in β-arbutin and kojic acid, indicating that the de-pigmenting effect of T1 is far more efficient than those of the high dose-dependent β-arbutin and kojic acid (Fig. 3). This result reveals that the T1 compound effectively reduces melanogenesis in zebrafish and is comparable with the melanin quantification in melanocytes.
As a desirable depigmenting agent, the natural compound T1 is highly safe and without any side effect. Acute oral toxicity test was carried out in the present study to evaluate the safety of T1. Throughout the 14 days of observation after oral administration, there were no deaths and clinical abnormal changes observed in the treated mice, which were dosed at 1500, 3000, 6000 mg of the T1 compound per kilogram of body weight. These treated mice did not show any signs of toxicity and the overall conditions of these mice were similar to that of the control ones. The toxicologically considerable changes in mean body weight were not observed in the animals treated with T1 (Supplementary Table S2 and Fig. 4). Thus, T1 exerted no adverse toxic effects in ICR mice at a dose up to 6000 mg/kg. The result of this study will be served as a reference of safety margin for human use.
Conclusion
This study screened 78 different Chinese herbal plants and identified the potent and functional components useful for tyrosinase inhibition, melanogenic inhibition and depigmentation. The natural component T1, isolated from Gastrodia elata, is a strong competitive inhibitor of mushroom tyrosinase (IC50 = 0.53 μM, Ki = 58 ± 6 nM), which is superior to kojic acid (IC50 = 40.69 μM) and more effective than other known natural products. The in vitro assay demonstrates that 50 μM of T1 apparently attenuates 20% melanin content of human normal melanocytes and shows no significant cell toxicity. The in vivo assay reveals that T1 effectively reduces melanogenesis in zebrafish without any adverse side effects. The acute oral toxicity study evidently confirms that the T1 molecule is free of discernable cytotoxicity in mice. The molecular models reveals that the sulfur atom of T1 interacting with the copper ions in the active site of tyrosinase is crucial for mushroom tyrosinase inhibition and the ability of disturbing the human melanin synthesis. In conclusion, T1 isolated from Gastrodia elata is a potential candidate in developing safe cosmetic and pharmacological agents, which is of great potency in skin-whitening.
References
Seo, S. Y., Sharma, V. K. & Sharma, N. Mushroom tyrosinase: recent prospects. J Agric Food Chem 51, 2837–2853, 10.1021/jf020826f (2003).
Prota, G. Progress in the chemistry of melanins and related metabolites. Med Res Rev 8, 525–556 (1988).
Spritz, R. A. & Hearing, V. J., Jr Genetic disorders of pigmentation. Adv Hum Genet 22, 1–45 (1994).
Lindquist, N. G. Accumulation of drugs on melanin. Acta Radiol Diagn (Stockh) 325, 1–92 (1973).
Solano, F., Briganti, S., Picardo, M. & Ghanem, G. Hypopigmenting agents: an updated review on biological, chemical and clinical aspects. Pigment Cell Res 19, 550–571, 10.1111/j.1600-0749.2006.00334.x (2006).
Briganti, S., Camera, E. & Picardo, M. Chemical and instrumental approaches to treat hyperpigmentation. Pigment Cell Res 16, 101–110 (2003).
Pawelek, J. M. & Korner, A. M. The biosynthesis of mammalian melanin. Am Sci 70, 136–145 (1982).
Passi, S. & Nazzaro-Porro, M. Molecular basis of substrate and inhibitory specificity of tyrosinase: phenolic compounds. Br J Dermatol 104, 659–665 (1981).
Solomon, E. I., Sundaram, U. M. & Machonkin, T. E. Multicopper Oxidases and Oxygenases. Chem Rev 96, 2563–2606 (1996).
Cabanes, J., Garcia-Canovas, F., Lozano, J. A. & Garcia-Carmona, F. A kinetic study of the melanization pathway between L-tyrosine and dopachrome. Biochim Biophys Acta 923, 187–195 (1987).
Cass, A. E. & Hill, H. A. Copper proteins and copper enzymes. Ciba Found Symp 79, 71–91 (1980).
Zhang, X., Hu, X., Hou, A. & Wang, H. Inhibitory effect of 2,4,2',4'-tetrahydroxy-3-(3-methyl-2-butenyl)-chalcone on tyrosinase activity and melanin biosynthesis. Biol Pharm Bull 32, 86–90 (2009).
Park, K. H. et al. Inhibitory effect of ammonium tetrathiotungstate on tyrosinase and its kinetic mechanism. Chem Pharm Bull (Tokyo) 54, 1266–1270 (2006).
Chen, Q. X. & Kubo, I. Kinetics of mushroom tyrosinase inhibition by quercetin. J Agric Food Chem 50, 4108–4112 (2002).
Jones, K., Hughes, J., Hong, M., Jia, Q. & Orndorff, S. Modulation of melanogenesis by aloesin: a competitive inhibitor of tyrosinase. Pigment Cell Res 15, 335–340 (2002).
Kubo, I. & Kinst-Hori, I. Flavonols from saffron flower: tyrosinase inhibitory activity and inhibition mechanism. J Agric Food Chem 47, 4121–4125 (1999).
Reish, O., Townsend, D., Berry, S. A., Tsai, M. Y. & King, R. A. Tyrosinase inhibition due to interaction of homocyst(e)ine with copper: the mechanism for reversible hypopigmentation in homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 57, 127–132 (1995).
Shimizu, K., Kondo, R. & Sakai, K. Inhibition of tyrosinase by flavonoids, stilbenes and related 4-substituted resorcinols: structure-activity investigations. Planta Med 66, 11–15, 10.1055/s-2000-11113 (2000).
Funasaka, Y., Komoto, M. & Ichihashi, M. Depigmenting effect of alpha-tocopheryl ferulate on normal human melanocytes. Pigment Cell Res 13 Suppl 8170–174 (2000).
Mishima, Y., Hatta, S., Ohyama, Y. & Inazu, M. Induction of melanogenesis suppression: cellular pharmacology and mode of differential action. Pigment Cell Res 1, 367–374 (1988).
Smith, C. J., O'Hare, K. B. & Allen, J. C. Selective cytotoxicity of hydroquinone for melanocyte-derived cells is mediated by tyrosinase activity but independent of melanin content. Pigment Cell Res 1, 386–389 (1988).
Maeda, K. F. M. In vitro effectiveness of several whitening cosmetic components in human melanocytes. J. Soc. Cosmet. Chem. 42, 361–368 (1991).
Arandt, K. A. F. T. B. Topical use of hydroquinine as a depigmenting agent. J. Am. Med. Assoc. 194, 117–119 (1965).
Fitzpatrick, T. B., Arndt, K. A., el-Mofty, A. M. & Pathak, M. A. Hydroquinone and psoralens in the therapy of hypermelanosis and vitiligo. Arch Dermatol 93, 589–600 (1966).
Kligman, A. M. & Willis, I. A new formula for depigmenting human skin. Arch Dermatol 111, 40–48 (1975).
Heilgemeir, G. P. & Balda, B. R. [Irreversible toxic depigmentation. Observations following use of hydroquinonemonobenzylether-containing skin bleaching preparations]. MMW Munch Med Wochenschr 123, 47–48 (1981).
Breathnach, A. C., Nazzaro-Porro, M., Passi, S. & Zina, G. Azelaic acid therapy in disorders of pigmentation. Clin Dermatol 7, 106–119 (1989).
Verallo-Rowell, V. M., Verallo, V., Graupe, K., Lopez-Villafuerte, L. & Garcia-Lopez, M. Double-blind comparison of azelaic acid and hydroquinone in the treatment of melasma. Acta Derm Venereol Suppl (Stockh) 143, 58–61 (1989).
Jimbow, K. N-acetyl-4-S-cysteaminylphenol as a new type of depigmenting agent for the melanoderma of patients with melasma. Arch Dermatol 127, 1528–1534 (1991).
Funayama, M. et al. Effects of alpha- and beta-arbutin on activity of tyrosinases from mushroom and mouse melanoma. Biosci Biotechnol Biochem 59, 143–144 (1995).
Kim, Y. J. & Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cellular and molecular life sciences: CMLS 62, 1707–1723, 10.1007/s00018-005-5054-y (2005).
Marrero-Ponce, Y. et al. Prediction of tyrosinase inhibition activity using atom-based bilinear indices. ChemMedChem 2, 449–478, 10.1002/cmdc.200600186 (2007).
Parvez, S. et al. Survey and mechanism of skin depigmenting and lightening agents. Phytother Res 20, 921–934, 10.1002/ptr.1954 (2006).
Curto, E. V. et al. Inhibitors of mammalian melanocyte tyrosinase: in vitro comparisons of alkyl esters of gentisic acid with other putative inhibitors. Biochem Pharmacol 57, 663–672 (1999).
Hermanns, J. F., Pierard-Franchimont, C. & Pierard, G. E. Skin colour assessment in safety testing of cosmetics. An overview. Int J Cosmet Sci 22, 67–71, 10.1046/j.1467-2494.2000.00021.x (2000).
Fuyuno, I. Spotlight turns on cosmetics for Asian skin. Nature 432, 938, 10.1038/432938a (2004).
Shimizu, K., Yasutake, S. & Kondo, R. A new stilbene with tyrosinase inhibitory activity from Chlorophora excelsa. Chemical & pharmaceutical bulletin 51, 318–319 (2003).
Park, S. H. et al. Terrein: a new melanogenesis inhibitor and its mechanism. Cellular and molecular life sciences: CMLS 61, 2878–2885, 10.1007/s00018-004-4341-3 (2004).
Zhong, S. et al. Depigmentation of melanocytes by the treatment of extracts from traditional Chinese herbs: a cell culture assay. Biol Pharm Bull 29, 1947–1951 (2006).
Choi, S. Y. et al. (4-Methoxy-benzylidene)-(3-methoxy-phenyl)-amine, a nitrogen analog of stilbene as a potent inhibitor of melanin production. Chem Pharm Bull (Tokyo) 50, 450–452 (2002).
Baek, S. et al. Inhibitory effect of dalbergioidin isolated from the trunk of Lespedeza cyrtobotrya on melanin biosynthesis. J Microbiol Biotechnol 18, 874–879 (2008).
Yanagihara, M. et al. Inhibitory effect of gnetin C, a resveratrol dimer from melinjo (Gnetum gnemon), on tyrosinase activity and melanin biosynthesis. Biol Pharm Bull 35, 993–996 (2012).
Roh, J. S., Han, J. Y., Kim, J. H. & Hwang, J. K. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol Pharm Bull 27, 1976–1978 (2004).
Kim, J. H., Kim, M. R., Lee, E. S. & Lee, C. H. Inhibitory effects of calycosin isolated from the root of Astragalus membranaceus on melanin biosynthesis. Biol Pharm Bull 32, 264–268 (2009).
Kong, Y. H. et al. Inhibitory effects of cinnamic acid on melanin biosynthesis in skin. Biol Pharm Bull 31, 946–948 (2008).
Cho, Y., Kim, K. H., Shim, J. S. & Hwang, J. K. Inhibitory effects of macelignan isolated from Myristica fragrans HOUTT. on melanin biosynthesis. Biol Pharm Bull 31, 986–989 (2008).
Lee, M. Y. et al. The melanin synthesis inhibition and radical scavenging activities of compounds isolated from the aerial part of Lespedeza cyrtobotrya. J Microbiol Biotechnol 20, 988–994 (2010).
Kim, J. P. et al. Melanocins A, B and C, new melanin synthesis inhibitors produced by Eupenicillium shearii. I. Taxonomy, fermentation, isolation and biological properties. J Antibiot (Tokyo) 56, 993–999 (2003).
Chen, L. G. et al. Melanogenesis inhibition by gallotannins from Chinese galls in B16 mouse melanoma cells. Biol Pharm Bull 32, 1447–1452 (2009).
Kim, S. J., Son, K. H., Chang, H. W., Kang, S. S. & Kim, H. P. Tyrosinase inhibitory prenylated flavonoids from Sophora flavescens. Biol Pharm Bull 26, 1348–1350 (2003).
Khan, S. B. et al. Tyrosinase-inhibitory long-chain esters from Amberboa ramosa. Chem Pharm Bull (Tokyo) 53, 86–89 (2005).
Nattapong, S. & Omboon, L. A new source of whitening agent from a Thai Mulberry plant and its betulinic acid quantitation. Nat Prod Res 22, 727–734, 10.1080/14786410601130794 (2008).
Hunt, K. J., Hung, S. K. & Ernst, E. Botanical extracts as anti-aging preparations for the skin: a systematic review. Drugs Aging 27, 973–985, 10.2165/11584420-000000000-00000 (2010).
Antignac, E., Nohynek, G. J., Re, T., Clouzeau, J. & Toutain, H. Safety of botanical ingredients in personal care products/cosmetics. Food Chem Toxicol 49, 324–341, 10.1016/j.fct.2010.11.022 (2011).
Reuter, J., Merfort, I. & Schempp, C. M. Botanicals in dermatology: an evidence-based review. Am J Clin Dermatol 11, 247–267, 10.2165/11533220-000000000-00000 (2010).
Baumann, L., Woolery-Lloyd, H. & Friedman, A. “Natural” ingredients in cosmetic dermatology. J Drugs Dermatol 8, s5–9 (2009).
Huang, N. K. et al. Neuroprotective principles from Gastrodia elata. J Nat Prod 70, 571–574, 10.1021/np0605182 (2007).
Kang, T. C. et al. Effect of vigabatrin on glutamate dehydrogenase in the hippocampus of seizure prone gerbils. Neurosci Lett 340, 115–118 (2003).
Kim, H. J., Moon, K. D., Oh, S. Y., Kim, S. P. & Lee, S. R. Ether fraction of methanol extracts of Gastrodia elata, a traditional medicinal herb, protects against kainic acid-induced neuronal damage in the mouse hippocampus. Neurosci Lett 314, 65–68 (2001).
Kim, H. J., Lee, S. R. & Moon, K. D. Ether fraction of methanol extracts of Gastrodia elata, medicinal herb protects against neuronal cell damage after transient global ischemia in gerbils. Phytother Res 17, 909–912, 10.1002/ptr.1246 (2003).
Huang, N. K., Lin, Y. L., Cheng, J. J. & Lai, W. L. Gastrodia elata prevents rat pheochromocytoma cells from serum-deprived apoptosis: the role of the MAPK family. Life Sci 75, 1649–1657, 10.1016/j.lfs.2004.05.008 (2004).
Hsieh, C. L. et al. Anticonvulsive and free radical scavenging activities of Gastrodia elata Bl. in kainic acid-treated rats. Am J Chin Med 29, 331–341, 10.1142/S0192415X01000356 (2001).
Hsieh, C. L. et al. Anticonvulsive and free radical scavenging activities of vanillyl alcohol in ferric chloride-induced epileptic seizures in Sprague-Dawley rats. Life Sci 67, 1185–1195 (2000).
Takahashi, M., Takara, K., Toyozato, T. & Wada, K. A novel bioactive chalcone of Morus australis inhibits tyrosinase activity and melanin biosynthesis in B16 melanoma cells. J Oleo Sci 61, 585–592 (2012).
Kumar, K. J., Yang, J. C., Chu, F. H., Chang, S. T. & Wang, S. Y. Lucidone, a novel melanin inhibitor from the fruit of Lindera erythrocarpa Makino. Phytotherapy research: PTR 24, 1158–1165, 10.1002/ptr.3018 (2010).
Kramer, H. J. et al. Malassezin, a novel agonist of the aryl hydrocarbon receptor from the yeast Malassezia furfur, induces apoptosis in primary human melanocytes. Chembiochem 6, 860–865, 10.1002/cbic.200400247 (2005).
Wang, K. H. et al. Cosmetic applications of selected traditional Chinese herbal medicines. J Ethnopharmacol 106, 353–359, 10.1016/j.jep.2006.01.010 (2006).
Choi, T. Y. et al. Zebrafish as a new model for phenotype-based screening of melanogenic regulatory compounds. Pigment cell research/sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society 20, 120–127, 10.1111/j.1600-0749.2007.00365.x (2007).
Wang, H. M., Chen, C. Y. & Wen, Z. H. Identifying melanogenesis inhibitors from Cinnamomum subavenium with in vitro and in vivo screening systems by targeting the human tyrosinase. Exp Dermatol 20, 242–248, 10.1111/j.1600-0625.2010.01161.x (2011).
Chen, Q. X., Ke, L. N., Song, K. K., Huang, H. & Liu, X. D. Inhibitory effects of hexylresorcinol and dodecylresorcinol on mushroom (Agaricus bisporus) tyrosinase. Protein J 23, 135–141 (2004).
Ghani, U. & Ullah, N. New potent inhibitors of tyrosinase: novel clues to binding of 1,3,4-thiadiazole-2(3H)-thiones, 1,3,4-oxadiazole-2(3H)-thiones, 4-amino-1,2,4-triazole-5(4H)-thiones and substituted hydrazides to the dicopper active site. Bioorganic & medicinal chemistry 18, 4042–4048, 10.1016/j.bmc.2010.04.021 (2010).
Veldman, M. B. & Lin, S. Zebrafish as a developmental model organism for pediatric research. Pediatr Res 64, 470–476, 10.1203/PDR.0b013e318186e609 (2008).
Shin, N. H. et al. Oxyresveratrol as the potent inhibitor on dopa oxidase activity of mushroom tyrosinase. Biochem Biophys Res Commun 243, 801–803, 10.1006/bbrc.1998.8169 (1998).
Lee, Y. C. et al. Phage Display-Mediated Discovery of Novel Tyrosinase-Targeting Tetrapeptide Inhibitors Reveals the Significance of N-Terminal Preference of Cysteine Residues and Their Functional Sulfur Atom. Molecular pharmacology, 10.1124/mol.114.094185 (2014).
Hsiao, N. W. et al. Serendipitous discovery of short peptides from natural products as tyrosinase inhibitors. Journal of chemical information and modeling 54, 3099–3111, 10.1021/ci500370x (2014).
Acknowledgements
We thank Mr. Bo-Chang Liao, Dr. Wen-Tai Li, Dr. Lie-Chwen, Lin (National Research Institute of Chinese Medicine, Ministry of Health and Welfare, Taipei, Taiwan), Chau-Lung Chen (Chuan Sheng Chinese Medicine Clinic, Taipei, Taiwan) and Hui-Min Wang (Department of Fragrance and Cosmetic Science, Kaohsiung Medical University, Kaohsiung, Taiwan) for fruitful discussions and help. We thanks the National Center for High-Performance Computing for computer time and facilities. The computations of GOLD and Discovery Studio were performed at the National Center for High-Performance Computing, Taiwan. This work was supported by National Research Institute of Chinese Medicine, Ministry of Health and Welfare [MM10211-0153] and Ministry of Science and Technology [Nos. MOST 100-2320-B-077-004-MY2 and 103-2320-B-077-001-MY3, awarded to K-C. T.], Taipei, Taiwan.
Author information
Authors and Affiliations
Contributions
W.C.C., T.S.T., N.W.H. and K.C.T. conceived the study. Y.L.L. provides T1 compound and performed acute oral toxicity assay in mice. Z.H.W. performed zebra-fish in vivo assay. T.S.T., Y.C.L. and H.H.L. performed mushroom tyrosinase inhibition assay, IC50 (Ki) determination, cell viability and Melanin quantification assay. W.C.C., T.S.T. and K.C.T. analyzed the data and contributed in manuscript writing. W.C.C., T.S.T., N.W.H., C.C.T. and K.C.T. made contributions in interpreting results, discussion of the associated molecular docking and improvement of this paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Data
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Chen, WC., Tseng, TS., Hsiao, NW. et al. Discovery of Highly Potent Tyrosinase Inhibitor, T1, with Significant Anti-Melanogenesis Ability by zebrafish in vivo Assay and Computational Molecular Modeling. Sci Rep 5, 7995 (2015). https://doi.org/10.1038/srep07995
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07995
This article is cited by
-
The Hull of Ripe Pistachio Nuts (Pistacia vera L.) as a Source of New Promising Melanogenesis Inhibitors
Plant Foods for Human Nutrition (2021)
-
Isolation of phenylpropanoid sucrose esters from the roots of Persicaria orientalis and their potential as inhibitors of melanogenesis
Medicinal Chemistry Research (2019)
-
Tyrosinase-based TLC Autography for anti-melanogenic drug screening
Scientific Reports (2018)
-
Tyrosinase inhibitory components from the seeds of Cassia tora
Archives of Pharmacal Research (2018)
-
Biological activities and biomedical potential of sea cucumber (Stichopus japonicus): a review
Fisheries and Aquatic Sciences (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
