
Abstract
The fungal microbiota is an important component of the human gut microbiome and may be linked to gastrointestinal disease. In this study, the fungal microbiota of biopsy samples from adenomas and adjacent tissues was characterized by deep sequencing. Ascomycota, Glomeromycota and Basidiomycota were identified as the dominant phyla in both adenomas and adjacent tissues from all subjects. Among the 60 genera identified, the opportunist pathogens Phoma and Candida represented an average of 45% of the fungal microbiota. When analyzed at the operational taxonomic unit (OTU) level, however, a decreased diversity in adenomas was observed and three OTUs differed significantly from the adjacent tissues. Principal Component Analysis (PCA) revealed that the core OTUs formed separate clusters for advanced and non-advanced adenomas for which the abundance of four OTUs differed significantly. Moreover, the size of adenomas and the disease stage were closely related to changes in the fungal microbiota in subjects with adenomas. This study characterized the fungal microbiota profile of subjects with adenomas and identified potential diagnostic biomarkers closely related to different stages of adenomas.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most common diagnosed cancer in males and the second most common diagnosed cancer in females worldwide; over 1.2 million colorectal cancer diagnoses and 608,700 colorectal cancer deaths were recorded in 20081. More than 80% of sporadic CRC cases were induced by colorectal adenoma2,3. Colorectal adenomas are classified into advanced or non-advanced stages according to the guidelines of the American Society for Gastrointestinal Endoscopy (ASGE). Advanced adenomas can further develop into carcinoma5. The gut microbiota has frequently been proposed to be associated with the pathogenesis of a range of diseases, including inflammatory bowel disease (IBD)6,7, diabetes8 and obesity9. A recent study indicated that the mucosa-adherent microbiota consisted of entrenched residents, such as Dorea spp., Faecalibacterium spp.10 and Fusobacterium spp.11,12,13, which might play a more direct role in the etiology of adenomas compared with its lumen counterpart by adjusting nutrient metabolism, providing competitive exclusion and stimulating the immune system14,15. However, fungi, as an important component of the intestinal microbiome, have been neglected because of their relatively low abundance and difficulty in culturing16. A study reported that the low abundance of gut microbes might alter the local microbiome, host immune responses and drive severe intestinal inflammation17. Therefore, the fungal gut microbiota must be extensively explored to obtain a clear picture of the natural communities and understand its role in the mucosal immune system that may lead to certain gut diseases, such as adenomas.
The fungal diversity in the human intestine has been assessed previously using denaturing gradient gel electrophoresis (DGGE) or clone library sequencing technology in healthy subjects and obese patients18,19,20,21. Only a small fraction could be detected and the fungal profile of intestinal samples could not be accurately represented. These limited results revealed that the fungal microbiota recovered in an individual was relatively homogenous (less than 10 phylotypes) and also suggested that eukaryotic communities were stable across time and were unique to individuals. In recent years, deep sequencing technologies have been introduced to characterize the fungal diversity in soil, human oral samples and fecal specimens of rodents, pig and dogs22,23,24,25,26. However, the human intestinal fungal microbiome has not been deeply investigated. Therefore, the human gut fungal microbiota should be examined with deep sequencing coverage to comprehensively understand the composition and distribution of fungal communities27. It is also very important to discover the specific pathogenic fungal species that might be involved in the etiology of gastrointestinal illness.
For colorectal adenomas, understanding the fungal agent associated with the etiology of advanced adenomas is crucial to evaluate the progress of adenomas from benign to highly dysplastic. In this study, fungal diversity in the adenomas and adjacent biopsy samples was characterized using a deep sequencing platform to reveal the structure of the human gut fungal microbiota and to determine whether a relationship exists between commensal or pathogenic fungi and the etiology of colorectal adenomas. These results might provide new biomarkers that could be used in the diagnosis of adenomas.
Results
Characteristics of the study subjects
In this study, we characterized the mucosa-adherent fungal microbiota of paired biopsy samples of adenomas and adjacent tissue from 27 subjects. The subjects were 56.3 years old on average and male subjects accounted for 63.0% of the population. Approximately two-thirds of the subjects (63.0%) were overweight and none of the subjects were obese. Nineteen subjects (19/27 = 70.0%) were classified with advanced stage disease; the remaining were classified with non-advanced subjects. Proximal and distal adenomas represented 48.1% and 51.9% of the sample population. Adenomas were categorized as small (1–5 mm; 3.7%), medium (6–10 mm; 81.5%) and large (>10 mm; 14.8%). The average number of adenomas per patient was 1.8 (range 1–6). The characteristics of the participants are presented in Table 1.
Taxonomy of the intestinal mucosal-associated fungal microbiota in subjects with adenomas
The intestinal fungal microbiota was characterized using an Illumina HiSeq 2000 platform combined with the fungal internal transcribed spacer (ITS) region ITS1 and ITS2 primer pair47. After quality filtration, 62,154,447 high quality reads were obtained from 54 intestinal biopsy samples, which resulted in an average of 1,151,008 reads per biopsy for taxonomy analysis. Table 2 presents the number of reads assigned to different level taxa and the percentage accounting for total qualified reads.
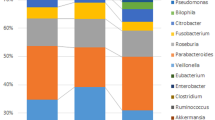
As shown in Figure 1A, the high-quality reads can be assigned to five fungi phyla (Supplementary Information Table 1). Three dominant phyla, Ascomycota (82.3% in adenomas and 80.5% in adjacent biopsy samples), Glomeromycota (3.6% and 3.1%, respectively) and Basidiomycota (2.5% and 2.6%, respectively), were present across all 54 specimens. Two rare phyla (relative abundance < 1%) were present: Chytridiomycota was present in 74% (40/54) of specimens and Neocallimastigomycota was present in 3.7% (2/54) of biopsy samples. Phylum Neocallimastigomycota is believed to come from diet and be transiently present in the human gut; therefore, it was excluded in the following analysis. A statistic comparison of the relative abundance of the phyla between adenomas and adjacent biopsy samples revealed that the phyla exhibiting the largest difference was Glomeromycota (p = 0.25).

Distribution of fungi in adenomas and adjacent biopsy samples at the phylum (Figure 1A), genus (Figure 1B) and species (Figure 1C) levels.
A: Adenoma biopsy; N: Normal biopsy. 1–27: case number of the biopsy samples.
We further analyzed the fungal microbiota structure of intestinal mucosa biopsy samples at the genus level. In total, there were 60 genera present in biopsy samples (Fig. 1B). Fifty-one genera were observed in adjacent mucosa specimens, whereas forty-four genera were present in adenomas (SI Table 1). Thus, a decreased diversity in adenomas was observed compared with adjacent biopsy samples. Intriguingly, among these genera, Phoma, an important opportunist pathogen, was dominant in all specimens and accounted for 54% and 39% of relative abundances in adenomas and adjacent biopsy samples, respectively. Candida was the other genus with a relatively high abundance present in adenomas (7%) and adjacent tissue samples (1%). These two genera accounted for 45% of the fungal microbiota in adenoma subjects. The low-abundance genera (0.01–1%) include Plectosphaerella, Cladophialophora, Cladosporium, Trichosporon, Rhodotorula and Thanatephorum. Furthermore, 36 species were discovered in studied biopsy samples (Fig. 1C). Excluding unclassified species, the species with the highest relative abundance was Candida tropicalis, which accounted for 3.6% and 2.1% of species in adenomas and adjacent biopsy samples, respectively. Other rare species (0.1–1%) included Rhodotorula glutinis and Cladosporium cladosporioides. However, no significant difference between adenomas and adjacent biopsy samples at the phylum, genus or species level were observed by t-test.
OTU-level analysis for the overall fungal microbiota composition of adenomas and paired adjacent biopsy samples
The qualified reads were clustered using CROP28 at 95% sequence identity for OTU generation. In total, there were 261 OTUs present in 54 mucosal biopsy samples (SI Table 1). After OTUs with less than 10 supported reads were removed, 232 OTUs remained. Ninety-one OTUs overlapped between adenomas and adjacent biopsy samples. Furthermore, 61 and 80 OTUs were uniquely present in adenomas and adjacent biopsy samples, respectively. Compared with adjacent biopsy samples, less diversity was observed in adenomas (Fig. 2A). Forty-eight core OTUs that were presented in more than 15% (8/54) of biopsy samples were selected for hierarchical clustering and PCA analysis to determine whether the core composition of the fungal microbiota can be separately clustered between adenomas and adjacent biopsy samples. Furthermore, for each biopsy, a rarefaction curve was drawn at a specific OTU level as a function of the observed number of OTUs on sequence counts at different sequencing depths. Figure 2B indicates that all rarefaction curves were saturated. Based on the core OTUs, the hierarchical clustering (SI Fig. 1A) and PCA (SI Fig. 1B) analysis revealed that adenomas and adjacent biopsy samples could not be classified into two separate clusters.

(Figure 2A). The OTU attribution in adenomas and adjacent mucosa biopsy samples. The blue circle indicates the OTUs present in adjacent biopsy samples and the brown circle indicates the OTUs present in the adenoma biopsy samples; overlap indicates the OTU shared by the two types of biopsy samples. (Figure 2B). Rarefaction curve demonstrating fungal sequence coverage in each biopsy. The curve depicts the number of OTUs observed at different sequencing depths where the x-axis is the number of sequences and the y-axis is the number of OTUs observed in each biopsy.
According to the etiology staging system, adenomas were classified into advanced or non-advanced stages. In this study, 8 subjects exhibited non-advanced disease and 19 subjects exhibited advanced adenoma. The hierarchical clustering and PCA analysis were conducted between subjects with advanced and non-advanced adenomas (Fig. 3A and Fig. 3B) and adjacent (SI Fig. 2A and SI Fig. 2B) biopsy samples. Compared with non-advanced biopsy samples, the hierarchical clustering analysis revealed that the advanced biopsy samples tended to cluster separately in both adenomas and adjacent biopsy samples (Fig. 3A and SI Fig. 2A). However, PCA analysis revealed such a tendency only in adenoma biopsy samples (Fig. 3B).

(Figure 3A). The heatmap and hierarchical clustering of subjects with advanced and non-advanced adenoma biopsy samples based on the core OTUs. Blue circles indicate the advanced biopsy samples; gray circles indicate the non-advanced biopsy samples. (Figure 3B). PCA analysis on advanced and non-advanced adenoma biopsy samples based on core OTUs. Red circles indicate the advanced adenomas; blue circles indicate the non-advanced biopsy samples.
The OTUs differed significantly between adenomas and adjacent biopsy samples or different stage biopsy samples
We selected OTUs with different abundances between adenomas and adjacent biopsy samples using a t-test and found that OTU 144089, which is assigned to phylum Basidiomycota, was significantly enriched in adjacent biopsy samples (p = 0.01) (Fig. 4A). The same test has also been conducted between adenomas and adjacent biopsy samples in advanced and non-advanced subjects. OTU 697566 (Fig. 4B), assigned to phylum Chytridiomycota, class Chytridiomycetes and order Spizellomycetales, was significantly enriched in adenomas compared with adjacent biopsy samples (p = 0.04); OTU 144089 (Fig. 4C) was significantly enriched in adjacent samples compared with adenomas in advanced subjects (p = 0.01). In non-advanced subjects, OTU 196869 (Fig. 4D), assigned to phylum Glomeromycota, class Glomeromycetes and order Paraglomerales, was significantly enriched in adenomas compared with adjacent biopsy samples (p = 0.04).

(Figure 4A). OTU 144089, which was assigned to phylum Basidiomycota, exhibited significant enrichment in adjacent tissues (p = 0.01) compared with adenoma biopsy samples in 27 paired biopsy samples. (Figure 4B). (OTU 697566), (4C) (OTU 144089) and (4D) (OTU 196869) demonstrate that the OTUs differed significantly between adenoma and adjacent biopsy samples in subjects with advanced and non-advanced disease. (Figure 4E). (OTU 5543) and (4F) (OTU 157510) demonstrate that the OTUs differed significantly in adenoma biopsy samples between subjects with advanced and non-advanced disease. (Figure 4G). (OTU 47507) and (4H) (OTU 339) indicate that the OTUs differed significantly in adjacent biopsy samples between subjects with advanced and non-advanced disease.
More importantly, there were two different clusters between advanced and non-advanced biopsy samples analyzed by PCA. Each of those clusters had two OTUs that may have acted independently in the development of adenomas. Statistical analysis revealed that two OTUs, 5543 (Fig. 4E; p = 0.01) and 157510 (Fig. 4F; p = 0.03), were significantly enriched in advanced adenoma biopsy samples compared with non-advanced adenoma tissue. OTU 5543 and OTU 157510 were assigned to order Saccharomycetales and phylum Basidiomycota. Furthermore, two OTUs, 47507 (4G; p = 0.02) and 339 (4H; p = 0.03), which were assigned to the Fusarium and Trichoderma genera, respectively, were significantly enriched in adjacent biopsy samples in advanced disease subjects compared with their counterparts in non-advanced subjects. Table 3 demonstrates that the OTUs differed significantly between adenomas and adjacent biopsy samples and between advanced and non-advanced biopsy samples.
To determine whether the structure of the fungal microbiota in the biopsy samples was correlated with the clinical data of the studied subjects, we compared the Shannon diversity of the samples with different clinical data using the abundance of core OTUs. The clinical data included adenoma size (diameter: 1–5 mm small; 6–10 mm media; >10 mm large), number of adenomas, body mass index (BMI) and disease stage (advanced and non-advanced). The relationship between the Shannon diversity and clinical data was evaluated using the Pearson coefficient. Table 4 demonstrates that adenoma size and disease stage were the most important clinical data and exhibited a close relationship with the OTU Shannon diversity.
Discussion
Despite extensive literature on the human gut microbiome, little is known regarding the gut fungal microbiota, especially the intestinal mucosal-associated mycobiota composition and dynamic changes during gut disease development. This lack of information may be the result of the low abundance of fungi in the gut microbiome or limited attention received from molecular analyses29,30. Fungi are an important part of the human gut microbiome and play both beneficial and harmful roles in humans30. Moreover, compared with the lumen counterpart, the mucosal associated fungal microbiota is more stable because the organisms adhere to surface-associated polysaccharide matrices of the gut epithelium31. Because the dysbiosis of intestinal mucosal-associated fungal microbiota may be associated with the etiology of IBD32 and CD33, changes in the gut fungal microbiota may be involved in the development of adenomas. In this study, we explored the profile of intestinal mucosal-associated fungal microbiota in 27 subjects with adenomas using the Illumina HiSeq 2000 platform. The results revealed that five fungi phyla were present in intestinal biopsy samples, including three dominant fungal phyla, Ascomycota, Glomeromycota and Basidiomycota and two rare phyla (relative abundance < 1%), Chytridiomycota and Neocallimastigomycota. Only two fungal phyla were reported in the fungal microbiota of intestinal biopsy samples from IBD patients, including Ascomycota and Basidiomycota32. The other study focusing on the gut mucosal mycobiota in CD reported that the main phyla were Saccharomycotina, Pezizomycotina and Basidiomycota33. The reason for this discrepancy among studies may be that different clinical samples were assayed and different methodologies (DGGE vs high throughput sequencing technology) were used.
Approximately 100,000 fungal species exist in the environment, but of these, only 300 species are known to cause animal or human infection34. Most of the fungal infections in humans are undiagnosed and under-reported35. In this study, sixty fungal genera were discovered and two opportunistic pathogenetic fungi genera, Phoma and Candida, were abundantly (45%) present in all 54 biopsy samples. Interestingly, Phoma only accounted for 2.8% of the fungi in oral rinse samples from healthy subjects21; however, this species was first observed as the predominant genus in intestinal biopsy samples of subjects with adenomas. As an opportunistic fungal pathogen, it has been reported that Phoma can cause lung mass36 and subcutaneous mycosis37,38 and therefore may also be involved in the formation of adenomas. In addition, Candida species, another opportunistic fungal pathogen22,39 that rarely colonizes the gastrointestinal tracts of healthy subjects40, were also discovered in our study at a relatively high abundance (2.8%) in all biopsy samples. Candida spp. are able to form biofilms, which makes the genera a potential cause of nosocomial infections35,41. Moreover, Candida tropicalis, perhaps associated with fungal infection in severe ulcerative colitis22, was also found to be present in all 54 biopsy samples at relatively high abundance. Taken all together, these results further proved that the pathogenesis of the dominant fungus in intestinal mycobiota may be common among patients with adenomas and is also most likely involved in the development of adenomas. Table 2 indicated that lower taxa were assigned fewer reads, indicating that the fungal database, especially the fungal database associated with humans, should be further expanded to provide more reference information for fungal mycobiota and to help identify potential fungi biomarkers in human gut illness.
We further explored the global structure of the fungal microbiota at the OTU level in biopsy samples of subjects with adenomas to determine whether individual OTUs are involved in the etiology of adenomas. In this study, 232 OTUs were identified in intestinal biopsy samples using high throughput sequencing technology. However, using DGGE and clone library sequencing, Ott et al.32 only found 43 OTUs in their study of intestinal mucosa-associated fungal microbiota in IBD patients and control subjects. Moreover, a decreased diversity of adenomas was observed compared with control biopsy samples in the present study. This result supported the hypothesis that fungal infection may play a role in chronic disease pathogenesis; however, an inverse correlation between fungal diversity and disease progression is more likely to occur because the microenvironment becomes less suitable for fungal growth, e.g., mucus dysfunction in cystic fibrosis (CF)42. At the global level, there were no significant differences between adenomas and adjacent biopsy samples. However, the individual OTU 144089, assigned to phylum Basidiomycota, was significantly enriched in adjacent biopsy samples compared with adenomas (p = 0.01) (Fig. 4A; Table 3). Furthermore, when comparing OTUs present in the adenomas and adjacent biopsy samples in advanced and non-advanced subjects, we found that OTU 144089 was also significantly enriched in adjacent samples compared with adenomas in advanced subjects (p = 0.01) (Fig. 4B; Table 3). Thus, OTU 144089 may be a commensal or beneficial OTU surviving in adjacent biopsy samples that is excluded by some factors, such as other fungal, bacteria or metabolic products. OTU 196869, assigned to phylum Glomeromycota, class Glomeromycetes and order Paraglomerales, was significantly enriched adenomas compared with adjacent biopsy samples in non-advanced subjects. Therefore, OTU 196869 may be involved in the development from normal biopsy samples to adenomas. OTU 697566, assigned to phylum Chytridiomycota, class Chytridiomycetes and order Spizellomycetales, was significantly enriched in adenomas compared with adjacent biopsy samples in advanced subjects (p = 0.04). Thus, OTU 697566 may be adaptive to the microenvironment of advanced adenomas, whereas OTU 196869 is better suited for the non-advanced adenomas microenvironment. Overall, the two enriched OTUs may be related to the presence of adenomas.
However, the detailed fungal microbiota composition of biopsy samples of different stages revealed that there was a discrepancy between advanced and non-advanced adenoma biopsy samples in PCA analysis (Fig. 3B). Furthermore, two OTUs, including OTU 5543 (p = 0.01) and OTU 157510 (p = 0.03), were significantly enriched in advanced biopsy samples compared with non-advanced adenomas tissue. OTU 5543 and 157510 were assigned to order Saccharomycetales and phylum Basidiomycota, respectively. This result indicated that the change of the fungal microbiota composition, especially with respect to OTU 5543 and 157510, may play a more important role in the development of non-advanced to advanced adenomas. However, we were unable to obtain further information regarding these OTUs because the sequences cannot be assigned to a lower level due to the absence of a reference in the ITS database. Furthermore, when compared with the adjacent biopsy samples between advanced and non-advanced subjects, two OTUs were observed to be significantly enriched in advanced subjects. One of the two important OTUs, OTU 47507, which can be assigned to genus Fusarium, was significantly (p = 0.02) enriched in advanced stages compared with non-advanced stages in adjacent biopsy samples. Some species of the Fusarium genus are believed to cause a range of infections in humans with normal or compromised immune systems because of the toxin it produces43. By altering the different intestinal defense mechanisms, cell proliferation, mucus layer, immunoglobulins (Ig) and cytokine production, Fusarium is therefore involved in infectious disease44. As one of the Fusarium toxins, Deoxynivalenol (DON) could play a role in diseases like inflammatory bowel disease (IBD)45,46. OTU 339, which was assigned to the Trichoderma fungal genus, has become a member of the emerging list of opportunistic pathogens. Trichoderma spp. has been reported to cause pulmonary mycetoma47, peritonitis48,49,50, infection of a perihepatic hematoma51 and disseminated disease52 in immunocompromised subjects. Therefore, Fusarium and Trichoderma colonization might be associated with the different stages of adenoma progression.
In this study, next-generation sequencing on the HighSeq platform combined with bioinformatics analyses were performed to investigate the intestinal fungal microbiota of subjects with adenomas. Our results revealed that there was a change in the fungal microbiota in adenoma patients at different stages of progression and that the opportunistic pathogenetic fungi predominant in mucosa biopsy samples may be a common profile in patients with adenomas. The OTUs with significant difference in biopsy samples might be associated with different stages of adenomas progression. However, because of ethical issues, we were unable to obtain biopsy samples from healthy individuals. Therefore, in the current study, we were unable to compare the adenoma biopsy samples with healthy biopsy samples. This sample should be included in future studies by recruiting healthy volunteers. Moreover, because of the variations in individual mycobiota, a large number of healthy biopsy samples should be collected to better define healthy mycobiota. Furthermore, the specific fungi of intestinal biopsy samples should be investigated to determine the fungi and the host interaction during the development of adenomas and whether the fungal microbiota is an etiologic factor of colorectal adenomas.
Methods
Study subjects and biopsy collection
Intestinal biopsy samples were collected from twenty-seven subjects, including seventeen men and ten women. All subjects were more than 50 years old and were undergoing a colonoscopy; visible adenomas were removed and sent to the pathology laboratory for histological examination. Adjacent rectal biopsy samples were collected in approximately 10–12 cm regions from the location of the adenomas. Biopsy samples were rinsed in sterile PBS, frozen in liquid nitrogen and stored at −80°C until use. Furthermore, these biopsy samples were categorized into two subgroups by a pathologist according to the American Society for Gastrointestinal Endoscopy (ASGE) Guideline: advanced (n = 19) and non-advanced (n = 8) stage adenomas4. Subjects did not use antibiotic or antifungal medication within the three months prior to the colonoscopy. Subjects had no known gastrointestinal disease in the previous year and no family history of CRC or other metabolic disease. All samples were collected in accordance with the relevant guidelines and regulations and the research was approved by the Research Ethics Boards of Beijing University People's Hospital. Documented informed consent was obtained from all participants.
DNA extraction and sequencing
Fungal genomic DNA was extracted from intestinal biopsy samples using the QIAamp DNA Stool Mini Kit (cat. No. 51504, Qiagen, Hilden, Germany) according to the manufacturer's instructions, with minor modifications. Briefly, biopsy samples were disrupted in an ASL buffer and were homogenized with 100 mg of zirconium beads (0.1 mm) in a Mini-Beadbeater-1 (Biospec Products Inc., Bartlesville, OK, USA) at a rate of 4800 rpm/min four times for 30 s each time at room temperature. Lysozyme was then added at a final concentration of 20 mg/mL (Sigma, St. Louis, MO, USA) and the suspension was incubated at 37°C for 40 min to improve lyses efficiency. Subsequently, the mixture was incubated in a 95°C water bath for 5 min to further increase the amount of total DNA extraction. Subsequent steps were performed according to the manufacturer's recommendations. A barcoded high-throughput sequencing library was prepared according to the method described by Caporaso et al.53 with an ITS1-2 primer pair. Briefly, the ITS region of the fungal DNA was amplified using the ITS1-F (5' CTTGGTCATTTAGAGGAAGTAA 3') and ITS2 (5' GCTGCGTTCTTCATCGATGC 3') primer pair54. In addition, to improve the fungus phylum coverage, the other four ITS-forward primers, each containing the same length as ITS1-F and starting from the ITS1-F 5' end adjacent nucleotide, were also used in the preparation of the sequencing library. For the other four forward primers, each 5' end nucleotide was designed by the aligned UNITE database using NCBI blast55. Both the forward and reverse primers had the appropriate Illumina adapters and sequencing primer pad. In addition, the reverse primer also contained a 6-bp barcode unique to each biopsy sample (SI Table 2). All PCR reactions were performed in 50-μl volumes containing 0.5 μM of each primer, 200 μM of each deoxynucleoside triphosphate dNTP mix, 2 mM MgCl2, 5 μl of 10×Taq polymerase buffer, 1 U Kapa Taq DNA polymerase (Kapabiosystems, Boston, US) and 1 μl (100 ng) genomic DNA. Reactions were held at 95°C for 5 min followed by 35 cycles of 95°C for 1 min, 53°C for 45 sec and 72°C for 1 min and a final elongation at 72°C for 7 min in an ABI thermocycler (Applied Biosystems 2720, USA). The PCR products were quantified with a Qubit 2.0 Fluorometer (Invitrogen). The fifty-four sequencing libraries were further quantified using real-time PCR on the ABI 7300 sequence detection system using the SYBR Green PCR Master Mix (Applied Biosystems). Paired-end sequencing (2 × 150 bp) was performed on an Illumina HiSeq 2000 sequencer in two lanes at the Center for Molecular Immunology of Chinese Academy of Sciences, Beijing, P.R. China.
Data analysis and taxonomic and OTU assignment
All raw reads were trimmed from both the 5′- and 3′-ends until 5 continuous bases with scores higher than 20 were found and the reads with shorter than 50 bp were removed. The high-quality read pairs with overlapping 3′-ends were merged and the non-overlapping pairs were concatenated using eight “N”s. Preprocessing was performed using HTQC56. For the OTU generation, both the overlapping and the non-overlapping reads were clustered using CROP28, which corresponds to a 95% sequence identity. The core OTUs were selected based on the criterion that the abundance was higher than 10 reads in at least 8 samples. The abundance profile of the core OTUs between the non-advanced and advanced-stage samples was compared. The abundance profile of the OTUs was generated using PCA analysis and the principle components of all samples were tested against adenoma size and number, body weight index (BMI) and disease stage (advanced and non-advanced). The representative sequences of the OTUs were aligned with the UNITE database57 using NCBI blast with an E value cutoff of 1e-5. The blast results were further filtrated by percent identity and the taxonomy lineages of the representative sequences were defined by the lowest common ancestor of the blast hits. The abundance profile of the samples was generated at the phylum, genus and species levels.
Statistical analyses
Sequence data were analyzed at several different scales. Fungal community composition was first examined at the phylum, genus and species levels and the overall differences in the fungal microbiota composition between adenomas and adjacent biopsy samples were evaluated using a paired t-test. The OTU composition between adenomas and adjacent biopsy samples was analyzed by hierarchical clustering using Spearman Ranked distance and PCA analysis. The OTUs with different abundances in the samples of advanced and non-advanced stages were identified using a two-tailed t-test. The relationships among fungal Shannon diversity, abundance and clinical data were evaluated using the Pearson Correlation.
Nucleotide sequence accession numbers
The sequence data of this study were submitted to the NCBI Sequence Read Archive (SRA) under accession number SRP045925.
References
Jemal, A. et al. Cancer statistics, 2010. CA Cancer J Clin 60, 277–300 (2010).
Jass, J. R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 50, 113–130 (2007).
Ullman, T. A. et al. Intestinal inflammation and cancer. Gastroenterology 140, 1807–1816 (2011).
Davila, R. E. et al. ASGE guideline: colorectal cancer screening and surveillance. Gastrointest Endosc 63, 546–557 (2006).
Terry, M. B. et al. Cancer Epidemiol Biomarkers Prev. Risk Factors for Advanced Colorectal Adenomas: A Pooled Analysis. Cancer Epidemiol Biomarkers Prev 11, 622–629 (2002).
DuPont, A. W. & DuPont, H. L. The intestinal microbiota and chronic disorders of the gut. Nat Rev Gastroenterol Hepatol 8, 523–531 (2011).
Carbonero, F. et al. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol 3, 448 (2012).
Qin, J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013).
Shen, X. J. & Rawls, J. F. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 1, 138–47 (2011).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22, 299–306 (2012).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22, 292–298 (2012).
McCoy, A. N. et al. Fusobacterium is Associated with Colorectal Adenomas. PloS One 8, e53653 (2013).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9, 313–323 (2009).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267 (2012).
Parfrey, L. W. et al. Microbial eukaryotes in the human microbiome: ecology, evolution and future directions. Front Microbiol 2, 153 (2011).
Rupnik, M. et al. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7, 526–536 (2009).
Scanlan, P. D. & Marchesi, J. R. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J 2, 1183–1193 (2008).
Walker, A. W. et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol 11, 7 (2011).
Hamad, I. et al. Molecular detection of eukaryotes in a single human stool sample from Senegal. PLoS One 7, e40888 (2012).
Gouba, N. et al. Plant and Fungal Diversity in Gut Microbiota as Revealed by Molecular and Culture Investigations. PloS One 8, e59474 (2013).
Ghannoum, M. A. et al. Characterization of the Oral Fungal Microbiome (Fungal microbiota) in Healthy Individuals. Plos Pathogen 6, e1000713 (2010).
Iliev, I. D. et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317 (2012).
Wang, H. et al. Patterns of human oral yeast species distribution on Hainan Island in China. Mycopathologia 76, 359–368 (2013).
Koh, A. Y. Gastrointestinal colonization of fungi. Curr Fungal Infect Rep 7, 144–151 (2013).
Bálint, M. et al. Illumina metabarcoding of a soil fungal community. Soil Biol Biochem 65, 128–132 (2013).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624 (2012).
Hao, X. et al. Clustering 16S rRNA for OTU prediction: a method of unsupervised Bayesian clustering. Bioinformatics 27, 611–618 (2011).
Huffnagle, G. B. & Noverr, M. C. The emerging world of the fungal microbiome. Trends Microbiol 21, 334–341 (2013).
Andersen, L. O. et al. Waiting for the human intestinal Eukaryotome. ISME J 7, 1253–1255 (2013).
Sonnenburg, J. L. et al. Getting a grip on things: how do communities of bacterial symbionts become established in our intestine? Nat Immunol 5, 569–573 (2004).
Ott, S. J. et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol 43, 831–841 (2008).
Li, Q. et al. Dysbiosis of Gut Fungal Microbiota is Associated Mucosal Inflammation in Crohn's Disease. J Clin Gastroenterol 00, 000–000 (2013).
Chandler, F. & Watts, J. [Mycotic, Actinomycotic and Algal Infections]. Anderson's Pathology Kissane J. (ed.), 391–432 (C.V. Mosby, St. Louis, Missouri, 1996).
V Ramana, K. et al. Invasive Fungal Infections: A Comprehensive Review. Am J Infect Dis Microbiol 1, 64–69 (2013).
Balis, E. et al. Lung mass caused by Phomaexigua. Scand J Infect Dis 38, 552–5 (2006).
De Hoog, G. S. et al. Atlas of Clinical Fungi 2 edn, 159–160 (Amer. Society for Microbiology, Washington, D.C., 2001).
Zaitz, C. et al. Subcutaneous Pheohypo-mycosis caused by Phoma cava. Report of a case and review of the literature. Rev Inst Med trop S Paulo 39, 43–48 (1997).
Mason, K. L. et al. Interplay between the gastric bacterial microbiota and Candida albicans during postantibioticre colonization and gastritis. Infect Immun 80, 150–158 (2012).
Douglas, L. J. Candida biofilms and their role in infection. Trends Microbiol 11, 30–36 (2003).
Peleg, A. Y. et al. Medically important bacterial-fungal interactions. Nat Rev Microbiol 8, 340–349 (2010).
Cui, L. et al. The human fungal microbiota in health and disease. Genome Med 5, 63075 (2013).
Schenk, M. & Mueller, C. The mucosal immune system at the gastrointestinal barrier. Best Pract Res Clin Gastroenterol 22, 391–409 (2008).
Antonissen, G. et al. The impact of Fusarium mycotoxins on human and animal host susceptibility to infectious diseases. Toxins 6, 430–452 (2014).
Maresca, M. & Fantini, J. Some food-associated mycotoxins as potential risk factors in humans predisposed to chronic intestinal inflammatory diseases. Toxicon 56, 282–294 (2010).
Maresca, M. From the gut to the brain: journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins 5, 784–820 (2013).
Escudero, M. R. et al. Micoma pulmonar causado por Trichoderma viridae. Actas Dermosifiliogr 67, 673–680 (1976).
Loeppky, C. B. et al. Trichoderma viridae peritonitis. South Med J 76, 798–799 (1983).
Ragnaud, J. M. et al. Infection periton'eale a ‘Trichoderma koningii sur dialyse p'eritoneale continue’ ambulatoire. Med Malad Infect 7, 402–405 (1984).
Tanis, B. C. et al. Fatal fungal peritonitis by Trichoderma longibrachiatum complicating peritoneal dialysis. Nephrol Dial Transplant 10, 114–116 (1995).
Jacobs, F. et al. Tricho- derma viridae infection in a liver transplant recipient. Mycoses 35, 301–303 (1992).
Gautheret, A. et al. Tricho- derma pseudokoningii as a cause of fatal infection in a bone marrow trans- plant recipient. Clin Infect Dis 20, 1063–1064 (1995).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Nat Acad Sci U S A 108, 4516–4522 (2011).
Bellemain, E. et al. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol 10, 189 (2010).
Abarenkov, K. et al. The UNITE database for molecular identification of fungi recent updates and future perspectives. New Phytol 186, 281–285 (2010).
Yang, X. et al. HTQC: a fast quality control toolkit for Illumina sequencing data. BMC Bioinformatics 14, 33 (2013).
Koljalg, U. et al. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol 166, 1063–1068 (2005).
Acknowledgements
The study was supported by grants from the China Ministry of Science and Technology Project 973 (numbers 2015CB554204), the National Natural Science Foundation of China (numbers 31471203 and 81401701) and the National Science and Technology Major Project (2013ZX10003002-002).
Author information
Authors and Affiliations
Contributions
C.L. and B.Z. designed and conceived the experiments. L.X., Y.Z. and N.W. collected the samples. C.L., H.M. and N.L. performed the experiments. X.Y. and X.X. analyzed data and prepared figures 1–4. B.Z. and C.L. wrote the manuscript text. Y.Z., Y.L., Y.H. and R.Z. reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Luan, C., Xie, L., Yang, X. et al. Dysbiosis of Fungal Microbiota in the Intestinal Mucosa of Patients with Colorectal Adenomas. Sci Rep 5, 7980 (2015). https://doi.org/10.1038/srep07980
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07980
This article is cited by
-
Plastiphily is linked to generic virulence traits of important human pathogenic fungi
Communications Earth & Environment (2024)
-
Intratumoural microbiota: a new frontier in cancer development and therapy
Signal Transduction and Targeted Therapy (2024)
-
Gut microbiota in colorectal cancer development and therapy
Nature Reviews Clinical Oncology (2023)
-
Broomcorn millet (Panicum miliaceum L.) tolerates soil salinity by regulating salt-tolerance mechanism and reshaping rhizosphere microorganisms
Plant and Soil (2023)
-
Emerging role of human microbiome in cancer development and response to therapy: special focus on intestinal microflora
Journal of Translational Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
