
Abstract
TRPM7 is an unusual bi-functional protein containing an ion channel covalently linked to a protein kinase domain. TRPM7 is implicated in regulating cellular and systemic magnesium homeostasis. While the biophysical properties of TRPM7 ion channel and its function are relatively well characterized, the function of the TRPM7 enzymatically active kinase domain is not understood yet. To investigate the physiological role of TRPM7 kinase activity, we constructed mice carrying an inactive TRPM7 kinase. We found that these mice were resistant to dietary magnesium deprivation, surviving three times longer than wild type mice; also they displayed decreased chemically induced allergic reaction. Interestingly, mutant mice have lower magnesium bone content compared to wild type mice when fed regular diet; unlike wild type mice, mutant mice placed on magnesium-depleted diet did not alter their bone magnesium content. Furthermore, mouse embryonic fibroblasts isolated from TRPM7 kinase-dead animals exhibited increased resistance to magnesium deprivation and oxidative stress. Finally, electrophysiological data revealed that the activity of the kinase-dead TRPM7 channel was not significantly altered. Together, our results suggest that TRPM7 kinase is a sensor of magnesium status and provides coordination of cellular and systemic responses to magnesium deprivation.
Similar content being viewed by others


Introduction
TRPM7 is an ubiquitously expressed protein that has an unusual structure: it contains both an ion channel and a protein kinase within a single polypeptide chain. TRPM7 is an essential gene and its knockout results in arrest of cell proliferation1,2 and early embryonic lethality in mice2,3. TRPM7 and its close homolog, TRPM6, are the only known channel kinases in vertebrates and both have been implicated in regulation of Mg2+ homeostasis (reviewed in ref. 4). TRPM7 and TRPM6 are known to form TRPM6/7 heterooligomers that could mediate relatively high magnesium currents in intestinal and kidney epithelia cells involved in mediation of magnesium uptake5,6. Several works showed that TRPM6 can form magnesium-permeable channels on its own7,8, however other studies suggested that TRPM6 can function only as a TRPM6/7 heterooligomer6.
TRPM7 ion channel domain belongs to a family of Transient Receptor Potential Melastatin-related (TRPM) channels (reviewed in refs. 9,10,11,12,13). The biophysical properties of TRPM7 are relatively well characterized4. A number of works have established TRPM7 as a divalent cation specific channel that is permeable to a number of physiologically important divalent cations, including Mg2+ and Ca2+, as well as to some toxic divalent cations. The TRPM7 channel is constitutively active and is regulated by free intracellular Mg2+ and Mg-ATP1,14,15. The biophysical properties of heterologously expressed TRPM7 are quite well understood; less is known about native TRPM7 (reviewed in ref. 4). Endogenous TRPM7-like currents have been detected in all cell types examined thus far5,6,16. A recent study found that endogenous TRPM7 currents assessed in human embryonic kidney cells (HEK-293) had an IC50 for intracellular Mg2+ comparable to heterologous systems17.
Mg2+ is an abundant intracellular cation that plays indispensible structural and functional roles in many cellular activities. The TRPM7 channel was suggested to provide a major mechanism of Mg2+ entry into the cell, thus regulating both cellular18 and whole body Mg2+ homeostasis2. Deletion of Trpm7 results in severe proliferation defects in DT-40 cells19 as well as in embryonic stem cells2, consistent with the fact that proliferating cells require Mg2+. Indeed, raising Mg2+ concentration in the growth medium fully rescues proliferation defects of Trpm7 mutant cells2,19, suggesting that the major role of TRPM7 is regulating Mg2+ intake. Consistent with the key role of TRPM7 in proliferation of most cell types, homozygous deletion of Trpm7 in mice causes early embryonic lethality2,3. In our previous work we showed that TRPM7 kinase domain-deficient (Δkinase) embryonic stem cells do not proliferate in regular medium containing 1 mM Mg2+ and their proliferation defect can be rescued by adding 10 mM Mg2+ to the medium2. These findings were recently confirmed in a study from another laboratory20. Significantly, Trpm7Δkinase/+ heterozygous mice are viable and develop Mg2+ deficiency that can be rescued by additional dietary Mg2+2.
The function of TRPM7 kinase is not well understood. The kinase domain of TRPM7 belongs to an atypical alpha-kinase family21. Alpha kinases do not display sequence similarity to conventional protein kinases and are able to phosphorylate residues within alpha –helices, while conventional kinases phosphorylate residues within unstructured and flexible regions22,23. The TRPM7 protein kinase domain is enzymatically active and has been shown to phosphorylate a number of substrates in vitro, including Annexin I and Myosin II24,25,26,27. However, the in vivo significance of these observations is not yet clear.
Since there is a good number of examples when the activity of a channel is regulated by phosphorylation28, the presence of a channel and a kinase within a single polypeptide chain of TRPM7 may suggest that the kinase activity influences the channel function. Indeed, some studies have suggested that the TRPM7 kinase could affect the channel, modulating its sensitivity to Mg2+ inhibition; however, other studies reported that TRPM7 kinase activity did not alter channel function, making the effect of kinase activity on the TRPM7 channel controversial (reviewed in ref. 4).
Initially it was suggested that TRPM7 kinase is required for channel activity; it was shown that in the absence of ATP the channel was inactive and could be activated by addition of ATP29. However, it was pointed out that in this work Na-ATP salt was used and therefore the stimulatory effect of ATP addition is most likely due to chelation of Mg2+ by ATP, resulting in the relieving of the channel inhibition by Mg2+30. Indeed later it was shown that TRPM7 lacking kinase activity has the same channel activity as the wild type TRPM7 channel in the absence of Mg2+18. Interestingly, this study also demonstrated a slightly decreased sensitivity of these channels to magnesium inhibition18. Subsequently, more detailed studies confirmed these observations suggesting that the kinase domain is involved in mediating the inhibition of TRPM7 channel activity by magnesium, serving as a binding site for Mg2+ and Mg-ATP31. Matsushita and colleagues showed that the phosphotransferase activity of the kinase domain was not required for channel function since cells expressing kinase-inactive TRPM7 displayed active TRPM7 channel currents similar to the ones in control cells. However, they also reported that abrogation of the kinase activity did not affect the channel sensitivity to the inhibition by Mg2+32. It should be noted that all these experiments were performed on cell lines overexpressing recombinant TRPM7.
To address the physiological role of TRPM7 kinase and to study it in vivo we constructed a knock-in mouse strain in which the kinase activity is abrogated by changing Lys-1646 to Arg in the ATP-binding site of TRPM7 kinase domain. Our results demonstrate that although TRPM7 kinase-dead mice have essentially normal phenotype and unaltered TRPM7 channel activity, they exhibit resistance to dietary Mg deprivation. Our findings suggest that TRPM7 kinase takes part in a signaling mechanism sensing magnesium status of the organism and as such TRPM7 kinase may play a role of a sensor for cellular and systemic Mg2+ status.
Results
Constructing the mouse strain with inactive TRPM7 kinase
To study the physiological role of TRPM7 kinase phosphotransferase activity we used a genetic approach: we generated a TRPM7 kinase-dead allele in which lysine 1646 was replaced with an arginine. Lysine 1646 is essential for Mg-ATP binding; its replacement with arginine was shown to result in a complete loss of a TRPM7 kinase activity32. The knock-in mice were produced using a gene-targeting vector technique, with further mouse colony generation. The targeting vector was constructed and integrated using an in vivo recombination strategy (Fig. 1). The resulting chimeric mice demonstrated germline transmission of the kinase-dead TRPM7 allele.

Generation of TRPM7K1646R/K1646R knock-in mice.
(A) Schematic representation of the endogenous Trpm7 locus and the targeting vector; homologous recombination strategy was used to engineer embryonic stem (ES) cells; the vector contained K1646R (KR) point mutation in exon 34 that replaced lysine residue at position 1646 to arginine. (B) DNA isolated from mouse tail biopsies of F1 heterozygous offspring was analyzed by PCR to confirm the presence of an introduced Neo-cassette; primer set A2/F3 amplified a fragment of 2.6 kb when the Neo-cassette was introduced by the targeting construct. 1 kb ladder was used as a reference (lane 1); lanes 2, 5 and 6 represent PCR products amplified from representative DNA samples of F1 offspring of mice that did not contain NEO-cassette; lanes 3 and 4 represent PCR products amplified from representative tail DNA samples of F1 offspring of mice that were targeted correctly and contained NEO-cassette. (C) Confirmation of point mutation: tail DNA sample from correctly targeted F1 representative mouse was amplified using primer set SQ1/UNI (lane 1); generated product of 1.75 kb size was used for further sequencing analysis to confirm the introduced point mutation. 1 kb ladder was used as a reference (lane 2); the expanded correctly targeted ES clone was used as a positive control (lane 3) and wild type ES clone was used as a negative control (lane 4). (D) Tail DNA samples of representative F2 offspring mice were analyzed to confirm the point mutation presence and Neo-cassette deletion: to genotype wild type (+/+), TRPM7K1646R/+ (KR/+) and TRPM7K1646R/K1646R (KR/KR) mice F4/R4 primer set was used to generate 640 b.p. product for knock-in allele and 440 b.p. product for wild type allele. Lanes 1 and 2 show PCR products amplified from tail DNA samples of representative TRPM7K1646R/K1646R mice; lanes 3 and 4 show PCR products amplified from tail DNA samples of representative wild type mice; lanes 5 and 6 show PCR products amplified from tail DNA samples of representative TRPM7K1646R/+ mice; 1 kb ladder was used as a reference (lane 7). (E) The PCR products from SQ1/UNI reaction shown in C (lane 1) were sequenced with SQ2 primer to demonstrate the presence of the K1646R mutation: T → C replacement and representative traces are shown for representative F1 offspring mouse.
TRPM7K1646R/K1646R mice have an essentially normal phenotype
Neither TRPM7K1646R/K1646R nor TRPM7K1646R/+ mice displayed any notable anatomical or physiological abnormalities. The mutant mice grew and gained weight normally during the first 9 months of their life (Fig. 2). However by 19 month of age TRPM7K1646R/K1646R mice exhibited slightly reduced weight compared to wild type controls (Fig. 2). Litter sizes were similar for mutant and wild type mice, ranging from 7 to 8 pups per litter. The pups developed normally at a normal growth rate, maturing at the age of 3–4 weeks. The mean life span for both mutant and wild type mice was 32 months when maintained on a regular diet. Finally, we did not observe any obvious behavioral differences between mutant and wild type animals.

The body weight of wild type and TRPM7K1646R/K1646R mice at different ages.
Open and shaded bars correspond to wild type and TRPM7 kinase-dead mice respectively. Values are means +/− the standard error of the mean. A Student's t-test was used to calculate p-values (n = 7–13 mice per group); P < 0.05 was considered significant. * Significantly different from wild type mice of the same age. The difference for the body weight of wild type and kinase-dead mice at 19 month of age was significant with P-value 0.005.
TRPM7K1646R/K1646R mice exhibited resistance to dietary Mg2+ deprivation
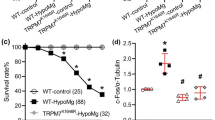
Since TRPM7 is implicated in the regulation of Mg2+ homeostasis, we hypothesized that the phenotype of the TRPM7 kinase-dead allele may be revealed specifically under conditions of Mg2+ deprivation. Thus, we placed the TRPM7K1646R/K1646R mutant and control wild type mice on Mg2+-depleted diet, which contained 0.003% of Mg2+. The onset of mortality for the wild type animals occurred at about 20 days of being placed on Mg2+-depleted diet. In contrast, the homozygous mutant mice started to die after about 60 days (Fig. 3). The median life span of homozygous mutant mice on a Mg2+-depleted diet increased approximately three-fold – from 2 months to 6 months compared to wild type mice. The maximal life span also increased. The life span of heterozygous mutants also increased compared to wild type mice though to a smaller extent than in homozygous mutants (Fig. 3).

K1646R mutation in TRPM7 confers resistance to Mg2+ deprivation in mice.
Animals carrying the TRPM7 kinase-dead allele showed improved survival on a Mg2+-deficient diet. The number of animals in was 20–22 per group of mice; each group contained equal numbers of males and females. The survival curves were significantly different with a log-rank (Mantel-Cox) test p-value < 0.02. The trends for the survival curves were also significantly different with a log-rank p-value of 0.005.
TRPM7K1646R/K1646R MEFs are resistant to Mg2+ deprivation
Next, we analyzed the ability of cells derived from the null mutant animals to adapt to low concentrations of Mg2+. We isolated mouse embryonic fibroblasts (MEFs) from wild type and homozygous mutant animals and compared their growth dynamics in regular (1 mM Mg2+) and Mg2+-depleted media. The two types of MEFs grew equally well in regular medium. However, when plated in Mg2+-depleted medium, the wild type MEFs exhibited transient growth arrest, while the growth of mutant MEFs was unchanged relative to their growth in 1 mM Mg2+ (Fig. 4). Thus, MEF's derived from kinase-dead mice were resistant to growth arrest caused by Mg2+ depletion. The Mg2+-depleted medium was prepared using a custom made Mg2+-free DMEM and dialyzed serum replacement. The residual Mg2+ concentration (i.e. Mg2+ that cannot be completely removed because it is tightly bound to serum proteins) was determined by atomic absorption spectrometry to be in the 10–20 μM range.

TRPM7K1646R/K1646R MEFs are resistant to Mg2+ deprivation.
Unlike WT cells, homozygous mutant MEFs proliferate equally well in the absence and presence of 1 mM Mg2+ in the growth medium. The results of experiment represent the average means of 3 experiments +− s.e.m.
Since TRPM7 can form heteroologomers with TRPM65,6 and TRPM7 kinase can trans-phosphorylate TRPM633, it is possible that TRPM6 may play a role in aforementioned observed effects of magnesium deprivation in MEFs. We performed RT (reverse transcriptase)-PCR and found that there was no expression of TRPM6 in both wild-type and TRPM7 kinase-dead mutant MEFs (see Supplementary materials Figure 1). Therefore, the observed effects of K1646R mutation on Mg2+ sensitivity in MEFs cannot be mediated by TRPM6.
Delayed-type of hypersensitivity response to oxazolone (allergic reaction)
It is known that low magnesium status conditions correlate with cell-extrinsic enhancement of systemic inflammatory and allergic responses34. Since TRPM7 kinase-dead mice displayed Mg2+ deprivation resistance we decided to evaluate their allergic reaction under magnesium deprivation conditions. Therefore, we evaluated the allergic reactions of TRPM7 kinase-dead animals by performing oxazolone sensitization experiments to evaluate the level of delayed-type hypersensitivity responses in mice.
One week prior to sensitization, wild type and TRPM7 kinase-dead mice were adapted to the control (0.2% Mg2+) diet. Under these conditions, TRPM7 kinase-dead mice displayed an oxazolone treatment sensitivity response similar to wild-type animals (Fig. 5). When the mice were placed on Mg2+-deficient diet, the wild type animals displayed an elevated allergic reaction to oxazolone treatment, while the response of the mutant mice to the treatment did not change (Fig. 5). When the mice were placed on a Mg2+-supplemented diet (0.3%), the hypersensitivity to oxazolone was fully abrogated in wild type mice, whereas TRPM7 kinase-dead mice were, again, unaffected. Therefore, Mg2+ -deficient diet does not enhance allergic response in TRPM7 kinase-dead mice. These results are consistent with increased survival of TRPM7 kinase-dead mice under the conditions of dietary Mg2+ deprivation. Taken together, our results demonstrate Mg2+ deprivation resistant phenotype of TRPM7 kinase-dead mice.

The effect of TRPM7 kinase deficiency and Mg2+ content in the diet on the oxazolone sensitivity reaction in mice.
The delayed sensitivity response is measured as a change in the ear thickness, mm. Before ear application of oxazolone wild type and TRPM7 kinase-dead mice were fed for 7 days a severely Mg2+ deficient, control and supplemental Mg2+ diet with 0.003, 0.1 and 0.3% Mg2+, respectively. The swelling response was measured 24 h after the challenge. Calculations of the swelling response were carried out comparing with the respective control groups not exposed to oxazolone. All mice were 4 months of age. Open and shaded bars correspond to wild type and TRPM7 kinase-dead mice respectively. Values are means +− s.e.m. Two-way of analysis of variant test was used for the calculations (n = 7–13-mice per group); P < 0.05 was considered significant; * significantly different from wild type mice fed a respective 0.003% Mg diet with P value < 0.01.
TRPM7K1646R/K1646R MEFs are oxidative stress resistant
Mg2+ deprivation is known to cause inflammation and elevate the levels of reactive oxygen species (reviewed in ref. 35), with the inflammatory response proposed to be responsible for oxidative damage during Mg2+ deficiency36. Since the kinase-dead mice displayed a significantly lower delayed-type response and sensitivity to oxazolone during Mg2+ deprivation compared to wild type animals, we explored the effect of loss of TRPM7 kinase activity on sensitivity to oxidative stress. To this end, MEFs isolated from wild type and TRPM7 kinase-dead mice were treated with hydrogen peroxide. After 1 hour of incubation the number of adhering wild type MEFs significantly decreased while the number of adhering TRPM7 kinase-dead MEFs did not change, suggesting that the TRPM7 kinase-dead cells were indeed more resistant to oxidative stress (Fig. 6).

TRPM7K1646R/K1646R MEFs are oxidative stress-resistant.
MEFs from kinase-dead mutant and wild type mice were grown in normal conditions and subsequently treated with 400 uM of hydrogen peroxide for 1 hour. Pictures were taken before and after 1 hour of hydrogen peroxide treatment. The density of the wild type MEFs after treatment is reduced compared to the KR mutant MEF cells.
Evaluation of Mg2+ status
The resistance of TRPM7 kinase-dead mice to dietary depletion of Mg2+ can arise either from undisturbed Mg2+ status or from an inability to sense an alteration in Mg2+ status. To assess Mg2+ status we analyzed Mg2+ concentration in blood serum of wild type and TRPM7 kinase-dead mice fed either standard or Mg2+-depleted diet. We found that Mg2+ concentration in the blood serum of wild type and TRPM7 kinase-dead animals was reduced to a similar extent by Mg2+ deprivation (Fig. 7A).

Assessment of Mg2+ status in TRPM7K1646R/K1646R mice.
The mice that were fed a control (0.2% Mg2+) diet and Mg2+-depleted (0.003% Mg2+) diet for 14 days prior to blood serum and bone sample preparation. (A) Mg2+ concentration in the blood serum of TRPM7 kinase-dead mutant and wild type mice was similarly reduced upon Mg2+ deprivation. (B) Mg2+ concentration in bones of the TRPM7 kinase-dead was reduced relative to wild type mice under conditions of regular Mg2+ but not sensitive to Mg2+ deprivation. Shaded and open bars correspond to wild type and TRPM7 kinase-dead mice respectively. Values are means +− s.e.m. Two-way of analysis of variance test was used for the calculations (n = 5–7 -mice per group); P < 0.05 was considered significant; * significantly different from wild type mice fed a respective 0.003% Mg diet with P value < 0.01. All mice were males 6 month of age.
The main physiological way to access Mg2+ during hypomagnesaemia is to utilize Mg2+ in bones, which represent the body's largest Mg2+ storage. Thus, we measured Mg2+ concentration in the bones of wild type and TRPM7 kinase-dead animals. We found that the wild type mice responded to Mg2+ deprivation as expected, i.e. by utilizing bone Mg2+ and thus lowering the Mg2+ concentration in their bones (Fig. 7B). Interestingly, in TRPM7 kinase-dead mice, under normal conditions bone Mg2+ concentration was decreased relative to controls and was insensitive to Mg2+ deprivation, remaining unchanged upon switch to a low Mg2+ diet (Fig. 7B). These results indicate that a deficit in dietary Mg2+ was not compensated by accessing bone Mg2+ in Mg2+ deprived TRPM7 kinase-dead mice, suggesting that TRPM7 kinase is necessary for sensing the lowered Mg2+ status.
Wild type and K1646R mutant TRPM7 channels have similar Mg2+ sensitivity in the physiological range
Electrophysiological analysis in heterologous overexpression systems has shown that the point mutation K1648R in human TRPM7 protein renders channel activity less sensitive to intracellular Mg2+ inhibition at low intracellular Mg2+ concentrations18. We therefore completed two full dose-response curves of TRPM7 to intracellular free Mg2+ in MEFs isolated from both wild type (WT) and TRPM7 K1646R mutant (KR) mice (Fig. 8A, B). WT currents had an IC50 of 71 ± 22 μM with a Hill coefficient of 0.7, where as TRPM7 KR mutants had an IC50 of 378 ± 127 μM with a Hill coefficient of 1.6 (fits not shown). Current amplitudes plateaued at 70 μM intracellular Mg2+ in the TPRM7 KR mutant, whereas WT had not reached plateau with Mg2+ concentrations as low as 25 μM and needed the complete removal of intracellular Mg2+ to reach the plateau phase (Fig. 8C). Normalized current amplitudes between the two mouse models at nominally free (0 μM) intracellular Mg2+ were statistically different with p < 0.05 as assessed by Student's T-test. A sigmoidal curve fit to the current development of individual cells from datasets in Fig. 8A and 8B was performed to assess the average maximal current amplitude, half-time to maximal current and rate of current development (see methods) at 0 μM, 70 μM and 210 μM intracellular free Mg2+ concentrations. This revealed that TRPM7 K1646R currents reached half-maximal current 70 s earlier than WT at 0 μM Mg2+ (182 s ± 36 s compared to WT with 252 s ± 21 s, n = 5 each), however there was no difference in the rate of current development (58 s ± 8 s compared to WT with 56 s ± 8 s). At 70 μM Mg2+, TRPM7 K1646R currents also reached half-maximal currents earlier than WT (150 s ± 20 s vs. 360 s ± 37 s, respectively; n = 5 each) and in addition showed faster rate of current development (43 s ± 5 s compared to WT with 97 s ± 21 s). At and above 210 μM intracellular free Mg2+ however, WT TRPM7 currents reached the half-maximal current point faster than the K1646R mutant currents (310 s ± 41 s vs. 537 s ± 185 s, respectively) and had a faster rate of current development (98 s ± 23 s vs. 233 s ± 76 s, respectively). None of the observed differences were statistically significant.

Mg2+ dose-response curve and divalent-free solution application on wild type (WT) and K1646R (KR) mutant TRPM7 currents in MEFs.
(A) Development of endogenous TRPM7 currents in wild type (WT) MEFs in response to increasing intracellular free Mg2+ concentrations ranging between 0 μM and 3200 μM as indicated by the symbols (n = 5–7). Currents were elicited by a voltage ramp ranging from −100 mV to +100 mV over 50 ms and given at 0.5 hz. Currents were extracted at +80 mV, normalized for cell size as pA/pF, averaged and plotted against time. Error bars are S.E.M. (B) Same conditions as in panel A, but assessed in TRPM7 KR mutant MEFs (n = 5–7). (C) Bar graph of averaged and normalized TRPM7 currents in WT and KR mutant MEFs. Data are taken from panels A and B displaying averaged current amplitude at 600 s experimental time and +80 mV. The star indicates statistical significance with p < 0.05. (D) Averaged endogenous TRPM7 current-voltage (I/V) relationship in WT and KR mutant MEFs in the absence of intracellular Mg2+ and extracted at 500 s (WT) or 400 s (KR) into the experiment (n = 5 each). Arrow indicates the voltage (+80 mV) where current amplitudes displayed in panel A and B were extracted. (E) Development of averaged and normalized endogenous TRPM7 currents in WT and KR mutant MEFs in absence of intracellular Mg2+ extracted at +80 mV over time. Voltage protocol and analysis as in panel A. Application of divalent-free external solution (see methods) at 400 s into the experiment, as indicated by the black bar (n = 7–9). Note the addition of 1 mM CsEDTA to the intracellular solution (see methods) to further reduce nominally free Mg2+ concentrations. (F) Average I/V relationship of endogenous WT (solid line) and KR mutant (dotted) TRPM7 currents extracted at experimental time 398 s (before application) and 450 s (at the end of application) (n = 7–9). Arrow indicates the voltage (+80 mV) where current amplitudes displayed in panel E were extracted.
To assess whether the amount of active channel proteins was similar in both mouse models, we kept the intracellular solution Mg2+ free and in addition applied a divalent-free extracellular solution to the fully developed TRPM7 current (see methods). This confirmed that there was no significant difference in the amount of TRPM7 channels in the plasma membrane between WT and TRPM7 K1646R mutant MEFs (Fig. 8E, F). We conclude that WT and TRPM7 K1646R mutant channels in MEFs have similar intracellular Mg2+ sensitivity, particularly at physiological Mg2+ levels.
Discussion
Our results demonstrate that inactivation of TRPM7 kinase affects the ability of the cells and the whole organism to sense and respond to Mg2+ deprivation. The mice lacking TRPM7 kinase activity survive severe dietary Mg2+ deprivation significantly better then the wild type mice.
It is known that Mg2+ deficiency status correlates with enhanced systemic inflammation and allergic responses34. In line with better Mg2+ deprivation survival of TRPM7 kinase-dead mice, they did not display elevated delayed -type hypersensitivity (allergic) response under severe dietary Mg2+ deprivation conditions. Similarly, on the cellular level TRPM7 kinase deficiency rendered MEFs resistant to Mg2+ deprivation: the growth rate of MEFs obtained from TRPM7 kinase-dead mice was not suppressed by Mg2+ depletion in the media while MEFs from the wild type mice displayed transient growth arrest.
Our results may have the following possible explanations: either loss of TRPM7 kinase activity improves the ability of cells and the whole organism to maintain the normal range of Mg2+ or it affects the ability to sense and respond to lowered Mg2+ concentrations. It is possible that kinase-dead mice can maintain Mg2+ homeostasis better than wild type animals under the conditions of Mg2+ deprivation. One of the important markers of Mg2+ deficiency in mammals is lowered Mg2+ in bones, which serve as a major Mg2+ storage in the body. Severe Mg2+-depleted diet caused a significant loss of Mg2+ from the bones of wild type mice and brought their Mg2+ bone concentrations to the similarly lower level as the ones we observed for TRPM7 kinase-dead mice. Also Mg2+ concentrations in the blood plasma under conditions of severe dietary Mg2+ deprivation were lowered to the same extent in both types of mice. Interestingly, TRPM7 kinase-dead mice have lowered Mg2+ concentrations in the bones while being on a regular diet. Therefore the better ability of TRPM7 kinase-dead mice to maintain Mg2+ homeostasis is unlikely. It is more plausible that abrogation of TRPM7 kinase activity leads to a loss of the ability of the cells and the whole organism to sense lowered Mg2+ status and to properly respond to it.
Since TRPM7 is a bi-functional protein and is implicated in cellular and systemic Mg2+ homeostasis (reviewed in ref. 4) it is possible that some effects of TRPM7 abrogated kinase activity may be due to TRPM7 altered channel function. While it is generally accepted that the kinase activity is not required for the channel gating, there is a number of studies that suggest that the kinase can modulate the activity of the channel. The question whether TRPM7 kinase can affect TRPM7 channel activity is controversial. In the systems using HEK cells overexpressing human TRPM7 it was demonstrated that the K1646R mutation leads to a decrease in channel sensitivity to Mg2+ inhibition18. However, in another study with the TRPM7 K1646R mutant overexpressed in HEK cells this effect was not observed32. Schmitz and colleagues18 found that both wild type and kinase-dead TRPM7 currents had similar amplitudes at 0 mM Mg2+ and they both were inhibited by 5 mM Mg2+. However, they found that at 3 mM Mg2+ TRPM7 kinase-dead mutant current was significantly higher than the wild type TRPM7 current. Matshushita and colleagues obtained similar results concerning the nonessential role of TRPM7 kinase activity for TRPM7 channel activation and mutant TRPM7 channel currents Mg2+ inhibition effects32. However, they did not find any significant difference in sensitivity to Mg2+ inhibition between wild type and K1646R mutant TRPM7 channel currents. The reason for the discrepancy is unknown; it may be related to differences in experimental procedures since Matsushita et al. used transient cell transfection while Schmitz et al. employed stable transfection for their experiments.
Another mechanism may be relied on ability of TRPM7 to modulate functions of other magnesium transporters. Among ~20 putative magnesium transporters/channels, TRPM6 is the most likely candidate for such modulatory mechanism. It was shown previously that TRPM6 and TRPM7 can form heteromeric channel complexes to regulate magnesium uptake by epithelial cells in the kidney and intestine (reviewed in refs. 11, 13). Furthermore, heterologous expression studies revealed that TRPM7 kinase can trans-phosphorylate TRPM633. Thus, it is possible that TRPM6/7 channel-kinase complexes lacking TRPM7 kinase activity do not respond properly to magnesium deprivation in TRPM7 kinase-dead mice. However, the effect of TRPM7 kinase inactivation on magnesium sensitivity in MEFs cannot be explained by TRPM6, since we found that TRPM6 is not expressed in MEFs (see Supplementary material, Figure 2).
In our experiments we found that endogenous currents in primary WT and TRPM7 kinase-dead MEFs are similarly sensitive to intracellular Mg2+. A difference in maximal current amplitude between WT MEFs and kinase-dead MEFs was observed for 0 μM intracellular Mg2+, but no difference was observed for all other Mg2+ concentrations, including the physiological range. Quantification of the time required to reach half-maximal current amplitudes and rates of current development showed increased kinetics for the TRPM7 KR mutant compared to TRPM7 WT at Mg2+ concentrations below 210 μM, however this was not statistically significant. Thus, absence of kinase phosphotransferase activity does not significantly alter TRPM7 channel behavior in MEFs in regards to intracellular Mg2+.
Disruption of phosphotransferase activity also does not seem to change the available number of TRPM7 channels in the plasma membrane, as evidenced by our DVF experiment (Fig. 8E, F). The slight difference in current behavior seen between Fig. 8A, B compared to 8E at 0 μM free Mg2+ could be due to the addition of EDTA to the intracellular solution for the DVF experiments (Fig. 8E). Chelating not only Ca2+ but also other divalent ion contaminants by EDTA might affect channel conformation and activity. Furthermore, in contrast to other cell types studied1,2, removal of divalent ions linearizes TRPM7 currents in MEFs, but does not significantly increase overall current amplitudes at depolarized potentials.
A recent study found that endogenous TRPM7 currents assessed in HEK-293 cells had an IC50 for intracellular Mg2+ comparable to heterologous systems, as did endogenous TRPM7 in SHEP (570 μM, 720 μM, 425 μM, respectively17). It is interesting to observe that native TRPM7 channels in primary MEFs seem more sensitive to intracellular Mg2+ than in the cell line HEK293 or SHEP-21N cell lines, which could point to different regulatory proteins determining overall channel activity in these diverse cell systems. Even in experimental hypomagnesemic condition intracellular Mg2+ is not expected to fall to less than a few hundred μM37,38, therefore it can reasonably be expected that TRPM7 activity is kept suppressed in MEF under normal circumstances, regardless of phosphotransferase activity. If TRPM7 channel Mg2+ dependence is similar in other cell types to the one observed in MEFs, it can be concluded that TRPM7 channel activity is unlikely to be significantly affected by its kinase activity and the effects observed in TRPM7 kinase-dead mice and MEFs obtained from them are not due to an altered channel function.
Very recently Kaitsuka et al. reported the generation of TRPM7 kinase-dead mice by introducing a K1646R point mutation39. They analyzed some aspects of phenotype of kinase-inactive mutant mice as well as electrophysiological properties of TRPM7 channels in cells derived form these mice39. In their experiments kinase-dead mice had normal blood serum total magnesium and calcium levels. The viability was normal, as well as food intake, body weight and locomotor activity. Our results are in general agreement with the results of Kaitsuka et al. In our experiments we also did not observe any differences in blood serum magnesium and viability. Although we observed no body weight differences up to the age of 9 months, 30% reduced body weight was observed for 19-month-old kinase-dead mutant mice. That difference was not observed by Kaitsuka et al, since they monitored the body weight for the first 10 months of animal's life. We also found significant differences in phenotypes of MEFs obtained from mutant and wild type animals under the conditions of magnesium deprivation. Electrophysiological studies in Kaitsuka et al. paper revealed that inactivation of TRPM7 kinase does not significantly affect the function of endogenous TRPM7 channels. The experiments were performed on peritoneal macrophages derived from TRPM7 kinase-dead mice. In our studies we performed electrophysiological experiments on MEFs and our results are in general agreement with that of Kaitsuka et al.
Our results demonstrate that in MEFs the abrogation of TRPM7 kinase activity does not significantly alter channel activity. It is possible that the previously observed effects of TRPM7 kinase inactivation on TRPM7 channel Mg2+ sensitivity were due to an indirect effect of additional cellular components that are absent in MEFs. Although TRPM7 kinase can autophosphorylate at multiple sites40 our results suggest that such phosphorylation is unlikely to be involved in the regulation of the channel activity. Our results also indicate that TRPM7 kinase is necessary for sensing the lowered Mg2+ concentrations since absence of the kinase activity leads to cell and organism insensitivity to lowered Mg2+ status, which in turn results in better survival of the organism under the conditions of Mg2+ deprivation.
Furthermore, since the kinase-dead mutant animals could survive significantly longer under conditions of Mg2+ deficiency than wild type animals, it can be suggested that death caused by Mg2+ deprivation is not due to insufficient amount of Mg2+per se (e.g. as required for some vital processes) but rather due to some toxic signaling pathways that are activated by Mg2+ deficiency via activity of TRPM7 kinase. Thus, our data suggest that physiological function of TRPM7 kinase is related to Mg2+ status and is part of a signal transduction mechanism that senses Mg2+ deprivation. The details of this mechanism still have to be elucidated. However, it is possible that TRPM7 kinase may be activated during Mg2+ deprivation, resulting in phosphorylation of various substrates, such as Annexin I and Myosin II, with a consequent cellular response such as growth arrest.
While our findings suggest that TRPM7 channel activity is not significantly influenced by its kinase activity, the possibility of kinase activity being dependent on channel activity cannot be excluded. Most likely, it is not a simple coincidence that channel domain and kinase domain have evolved to be encoded in one single molecule. TRPM7 channel-kinase is localized on the membrane and its kinase domain is kept anchored in the close proximity to the membrane and the channel pore. TRPM7 kinase is Mg2+-dependent and is activated by Mg2+ through Mg –ATP41. It is also possible that TRPM7 kinase can be activated through direct binding of Mg2+ to the kinase catalytic domain with subsequent changes in kinase domain conformation as it was shown for another member of the alpha-kinase family42. Since TRPM7 channel currents can be facilitated or inhibited by changes in Mg2+ concentrations1, the changes in local concentration of Mg2+ provided by influx through the channel pore may in turn induce conformational changes in the kinase domain with subsequent opening or closing of the kinase active site therefore making TRPM7 kinase domain acting as Mg2+ concentration sensor. It is possible that under conditions of Mg2+ deprivation TRPM7 channel is activated and its amplified currents create higher Mg2+ concentrations around the channel pore thus activating TRPM7 kinase; TRPM7 kinase passes the signal downstream by means of phosphorylation of various substrate proteins thus providing a response to magnesium deprivation. In such a case the absence of TRPM7 kinase activity would lead to the absence of normal response to Mg2+ deprivation as we observed in our experiments.
In our previous work we showed that deletion of the entire kinase domain of TRPM7 gene in mice results in early embryonic lethality while heterozygous mice survived and exhibited altered TRPM7 currents and hypomagnesaemic phenotype2. However, deletion of kinase domain most likely resulted in an altered overall structure of the protein, significantly altering channel pore structure that results in channel inactivation. Thus, deletion of full kinase domain can have an effect similar to a deletion of the entire TRPM7 gene. Indeed, our results with both types of knockout mice (knockout of the entire gene and deletion of the kinase domain) reveal similar effects on TRPM7 currents and similar early embryonic lethality (Ryazanova L.V; manuscript in preparation).
Mg2+ plays a critical physiological and pathological role in the organism. Mg2+ deprivation can be regarded as a particular type of stress: a reduction in serum Mg2+ levels leads to a number of potentially fatal health problems, including defects in cardiovascular system, motor function and behavior (reviewed in refs. 35, 43). It has been demonstrated that Mg2+ deprivation results in elevation of reactive oxygen species and subsequent oxidative stress34,36,44,45,46,47,48. Since we demonstrated that kinase-dead MEFs are resistant to Mg2+ deprivation and oxidative stress, it is tempting to suggest that during Mg2+ deprivation TRPM7 kinase senses oxidative stress and participates in the ensuing cellular response. It is possible that the TRPM7 kinase can act as Mg2+ sensor and as such can provide a response of the cell and the organism to a stressful Mg2+ deprivation. It is likely that TRPM7 kinase role in Mg2+ deprivation and oxidative stress response is a part of a global TRPM7 role in various types of stress responses of the cell and the organism.
Recent studies revealed the role of TRPM7 in various pathophysiological processes including cardiovascular diseases, hypertension, global ischemia and neuronal cell death (reviewed in ref. 4). In particular it was shown that oxygen-glucose deprivation-induced neuronal death requires TRPM7; it was furthermore suggested that activation of TRPM7 by reactive oxygen species could cause this effect49. In the mouse model of global ischemia, suppression of TRPM7 by small interfering RNA protected neurons in the hippocampus from cell death50. In these studies the involvement of TRPM7 kinase in cell death was not investigated. Our results suggest that TRPM7 kinase may be involved in cell death caused by oxidative stress and therefore could contribute to the effects observed in these studies. Further research in this direction can investigate the exciting potential of developing TRPM7 kinase inhibitors as drugs that protect cells and tissues from oxidative stress during various pathological conditions, for example during ischemia.
To our knowledge this work presents the first report of the physiological function of the TRPM7 kinase activity and demonstrates its essential role in cellular and systemic response to Mg2+ deprivation.
Methods
Animal studies
All animal procedures were carried out in accordance with the approved guidelines. The procedures were approved by Rutgers Robert Wood Johnson Medical School Animal Care and Use Committee (Protocol # I12-043-7).
Construction of TRPM7K1646R/K1646R kinase-dead mice
The targeting strategy is schematically outlined in Figure 1 and is described in detail below. The targeting vector was constructed by using long homology arm (LA) that extended from 5.31 kb 5′ to exon 34 and the LoxP/FRT flanked Neo- cassette. It was inserted in 1.06 kb 3′ end and A (4937) → G point mutation was engineered into exon 34. The short homology arm (SA) extended 2.17 kb 3′ to the LoxP flanked Neo-cassette.
Targeted iTl IC1(C57BL/6) embryonic stem cells were microinjected into Balb/c blastocysts. Resulting chimeras with a high percentage of a black coat color were mated to wild-type C57BL/6 mice to generate F1 heterozygous offspring. Mouse tail DNA was analyzed using the following primer set and PCR conditions to determine the presence of the Neo-cassette:
A2: 5′- GTCAAGGAACTACCTGGGAGAGG
F3: 5′- GCATAAGCTTGGATCCGTTCTTCGGA
F3 anneals inside the Neo cassette and A2 anneals 3′ to the short homology arm, outside the region used to create the targeting construct. A2/F3 amplifies a fragment of 2.6 Kb in length. PCR conditions were:
Hot Start 99C for 5 minutes
77C unlimited
1 cycle
Cycling Steps 94C for 20 seconds
62C for 60 seconds
72C for 2 minutes 30 seconds
32 cycles
Cooling Step 4C unlimited
Tail DNA samples from correctly targeted F1 mice were amplified by primer pair set SQ1and UNI and sequenced using primer SQ2 to confirm the introduced point mutation.
SQ1: 5-CAAGTAGCACTGGGTTACCGTTGC
UNI: 5′- AGCGCATCGCCTTCTATCGCCTTC
SQ2: 5′-GGAAACTCAGATCACAGCTTACAGTC
PCR conditions were:
Hot Start 99C for 5 minutes
1 cycle
Cycling Steps 94C for 20 seconds
60C for 30 seconds
72C for 2 minutes
32 cycles
72C for 7 minutes
Cooling Step 4C unlimited
SQ1/UNI amplifies a fragment of 1.75 Kb in length.
The PCR products from the SQ1/UNI reaction were sequenced with SQ2.
T → C point mutation was confirmed to be present in exon 34.
F1 heterozygous mice with K1646R point mutation and Neo-cassette included were crossed with FLP transgenic mice obtained from Jackson Lab (stock#005703) to delete the Neo-cassette and produce TRPM7K1646R/+ heterozygous (F2) mice with point mutation introduced and Neo-cassette deleted.
These F2 mice were genotyped using the following primer set and PCR condition:
F4 5′-TGTAACTTTCCTGCCCGAGTCT
R4 5′-CCGAGTTTCTGGAAGCCATG
94C 5 mins
94C 30 secs
62C 30 secs
72C 1 min
30 cycles
72C 10 mins
4C forever
For the wild-type allele the size of PCR product was about 440 bp. If Neo-cassette deletion occurred and point mutation was present in the allele, the size of the PCR product was about 640 bp.
TRPM7K1646R/+ F2 mice with introduced point mutation and deleted Neo- cassette were crossed with C57BL/6 mice to produce F3 generation of mice with removed FLP-transgene.
To detect the FLP-transgene in F3 mice, the following set of primers and PCR conditions were used:
FLP1: 5′-CACTGATATTGTAAGTAGTTTGC
FLP2: 5′-CTAGTGCGAAGTAGTGATCAGG
95°C 10 mins
95°C 20 secs
55°C 60 secs
72°C 60 secs
28 cycles
PCR product size was 725 bp when FLP-transgene was present.
TRPM7K1646R/+ F3 mice with introduced point mutation and absent both Neo-cassette and FLP-transgene were intercrossed to generate homozygous mice carrying the desired point mutation. Primer sets and PCR condition were the same as described above.
For PCR reactions Platinum Tag DNA polymerase and DNTP mix from Invitrogen were used.
All PCR products were separated on 1% agarose gels and stained with ethidium bromide.
The PCR product for confirming the point mutation (Figure 1C, lane 1) was cut from the gel, extracted using Qiagen Gel Extraction kit and submitted for sequencing.
Mouse diets and treatment
Mice were kept on 12/12-h light/dark cycle and allowed ad libitum access to food and water. The following diets were used for the experiments:
Magnesium deficient diet - 0.0015–0.003% of Mg, Teklad TD.93106,
Control (regular) diet - 0.2% of Mg, Teklad TD.07156.
Magnesium supplemented diet - 0.3% Mg, Teklad TD.120574.
Isolation and culture of primary mouse embryonic fibroblasts
TRPM7 kinase-dead MEF cells were obtained from E13.5dpc embryos as previously described2. All necessary reagents for preparation of MEF cell culture and performing the experiments were obtained from Invitrogen. Knockout DMEM without magnesium was custom made by Invitrogen. The absence of Mg2+ in the media was confirmed by Atomic Absorption Spectroscopy analysis. To prepare Mg2+-deficient DMEM dialyzed Knockout Serum Replacement was used. Knockout Serum Replacement was dialyzed using Slide-A-Lyzer Dialysis Cassette G2 (3,500 MWCO) from Thermo scientific. Knockout serum replacement was chosen to avoid variations in the quality of different batches of regular FBS. Residual magnesium level was determined by Atomic Absorption Spectroscopy analysis and was in 10–15 μM range which represented Mg2+ that could not be completely removed because it was tightly bound to a serum proteins).
Delayed-hypersensitivity response measurements
Oxazolone was used as a contact-sensitizing agent. Oxazolone (0.5%) dissolved in acetone was applied on the shaved mouse abdomen immediately before placing mice on the experimental diets with various Mg2+ content. Control animals were exposed to vehicle alone. Six days after sensitization, mice were challenged by epicutaneous application of 1% oxazolone in acetone on their ears. The swelling response was measured 24 h after challenge using an engineer's micrometer. Calculations of the swelling response were carried out by comparing to the respective control groups with no exposure to oxazolone.
Determination of total magnesium in the blood serum and bones
Blood samples were obtained from the hearts of 6-month old males. Blood serum was isolated by centrifugation at 1.2 g, 15 min at RT at stored at −80°C. Right tibias were dissected and cleaned from the muscle tissues and storied at −20°C. Total magnesium levels were determined using inductively coupled plasma-sector field mass spectrometry (ALS laboratories, Sweden) as described previously51.
Electrophysiology
Patch clamp experiments were performed in the whole-cell configuration. Currents were elicited by a ramp protocol from −100 mV to +100 mV over 50 ms acquired at 0.5 Hz and a holding potential of 0 mV. Inward current amplitudes were extracted at −80 mV, outward currents at +80 mV and plotted versus time. Data were normalized to cell size as pA/pF. Capacitance was measured using the automated capacitance cancellation function of the EPC-9 (HEKA, Lambrecht, Germany). Average cell capacitance for wild type (WT) MEF cells assessed at 0 mM Mg2+ was 26.4 pF (n = 5). Average cell capacitance for K1646R mutant MEF under the same conditions as WT was 23.5 pF (n = 5). Values over time were normalized to the cell size measured immediately after whole-cell break-in. Standard extracellular solution contained (in mM): 140 NaCl, 1.5 CaCl2, 2.8 KCl, 10 HEPES-NaOH, 0.5 Na-EDTA, 11 Glucose (pH 7.2, 300 mOsm). Divalent-free (DVF) extracellular solution contained (in mM): 140 NaCl, 2.8 KCl, 0 MgCl2, 10 HEPES-NaOH, 1 Na-EDTA, 11 Glucose (pH 7.2, 300 mOsm). Intracellular solution contained (in mM): 120 Cs-glutamate, 8 NaCl, 10 HEPES-CsOH, 10 Cs-BAPTA and appropriate amount of MgCl2 was added, as calculated with WebMaxC (Chris Patton, Stanford) (pH 7.2, 300 mOsm). The intracellular solution for the divalent free (DVF) extracellular solution experiment contained (in mM): 120 Cs-glutamate, 8 NaCl, 10 HEPES-CsOH, 10 Cs-BAPTA, 1 Na-EDTA (pH 7.2, 300 mOsm). A dose response curve fit was performed on wild type and K1646R mutant TRPM7 currents extracted from cells perfused with increasing Mg2+ concentrations using the following fit function: I = (Imax *(1/(1 + (IC50/x)ˆn))); where I is current, Imax is the maximal current amplitude, IC50 is half maximal inhibitory concentration and n is the Hill coefficient. Development of outward currents was quantitated for maximal current size, time to half-maximal current and rate of current development using a sigmoidal curve fit function with I = IBase + (Imax/(1 + exp((t1/2 − x)/τ))); where I is current, IBase is the baseline current, Imax is the maximal current amplitude, exp is exponent, t1/2 is the time to half-maximal current and τ is the rate of current development. Statistical analysis was performed using Student's t-test.
References
Nadler, M. J. et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595 (2001).
Ryazanova, L. V. et al. TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat Commun 1, 109 (2010).
Jin, J. et al. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760 (2008).
Fleig, A. & Chubanov, V. Trpm7. Handb Exp Pharmacol 222, 521–546 (2014).
Chubanov, V. et al. Hypomagnesemia with secondary hypocalcemia due to a missense mutation in the putative pore-forming region of TRPM6. J Biol Chem 282, 7656–7667 (2007).
Chubanov, V. et al. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A 101, 2894–2899 (2004).
Li, M., Jiang, J. & Yue, L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol 127, 525–537 (2006).
Voets, T. et al. TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 279, 19–25 (2004).
Runnels, L. W. TRPM6 and TRPM7: A Mul-TRP-PLIK-cation of channel functions. Curr Pharm Biotechnol 12, 42–53.
Chubanov, V., Gudermann, T. & Schlingmann, K. P. Essential role for TRPM6 in epithelial magnesium transport and body magnesium homeostasis. Pflugers Arch 451, 228–234 (2005).
Chubanov, V., Mederos y Schnitzler, M., Waring, J., Plank, A. & Gudermann, T. Emerging roles of TRPM6/TRPM7 channel kinase signal transduction complexes. Naunyn Schmiedebergs Arch Pharmacol 371, 334–341 (2005).
Dietrich, A., Chubanov, V. & Gudermann, T. Renal TRPathies. J Am Soc Nephrol 21, 736–744.
Schlingmann, K. P., Waldegger, S., Konrad, M., Chubanov, V. & Gudermann, T. TRPM6 and TRPM7--Gatekeepers of human magnesium metabolism. Biochim Biophys Acta 1772, 813–821 (2007).
Li, M. et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem 282, 25817–25830 (2007).
Monteilh-Zoller, M. K. et al. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol 121, 49–60 (2003).
Mederos y Schnitzler, M., Waring, J., Gudermann, T. & Chubanov, V. Evolutionary determinants of divergent calcium selectivity of TRPM channels. FASEB J 22, 1540–1551 (2008).
Zhang, Z. et al. The TRPM6 kinase domain determines the Mg.ATP sensitivity of TRPM7/M6 heteromeric ion channels. J Biol Chem 289, 5217–5227 (2014).
Schmitz, C. et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114, 191–200 (2003).
Sahni, J., Tamura, R., Sweet, I. R. & Scharenberg, A. M. TRPM7 regulates quiescent/proliferative metabolic transitions in lymphocytes. Cell Cycle 9, 3565–3574 (2010).
Krapivinsky, G., Krapivinsky, L., Manasian, Y. & Clapham, D. E. The TRPM7 Chanzyme Is Cleaved to Release a Chromatin-Modifying Kinase. Cell 157, 1061–1072 (2014).
Ryazanov, A. G. Elongation factor-2 kinase and its newly discovered relatives. FEBS Lett 514, 26–29 (2002).
Pinna, L. A. & Ruzzene, M. How do protein kinases recognize their substrates? Biochim Biophys Acta 1314, 191–225 (1996).
Ryazanov, A. G., Pavur, K. S. & Dorovkov, M. V. Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol 9, R43–45 (1999).
Clark, K. et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J 25, 290–301 (2006).
Clark, K. et al. The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett 582, 2993–2997 (2008).
Clark, K. et al. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J Mol Biol 378, 790–803 (2008).
Dorovkov, M. V. & Ryazanov, A. G. Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem 279, 50643–50646 (2004).
Ismailov, II & Benos, D. J. Effects of phosphorylation on ion channel function. Kidney Int 48, 1167–1179 (1995).
Runnels, L. W., Yue, L. & Clapham, D. E. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291, 1043–1047 (2001).
Fleig, A. & Penner, R. The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol Sci 25, 633–639 (2004).
Demeuse, P., Penner, R. & Fleig, A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol 127, 421–434 (2006).
Matsushita, M. et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J Biol Chem 280, 20793–20803 (2005).
Schmitz, C. et al. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J Biol Chem 280, 37763–37771 (2005).
Malpuech-Brugere, C. et al. Inflammatory response following acute magnesium deficiency in the rat. Biochim Biophys Acta 1501, 91–98 (2000).
Weglicki, W. B. Hypomagnesemia and inflammation: clinical and basic aspects. Annu Rev Nutr 32, 55–71 (2012).
Rayssiguier, Y., Durlach, J., Gueux, E., Rock, E. & Mazur, A. Magnesium and ageing. I. Experimental data: importance of oxidative damage. Magnes Res 6, 369–378 (1993).
Fujise, H., Cruz, P., Reo, N. V. & Lauf, P. K. Relationship between total magnesium concentration and free intracellular magnesium in sheep red blood cells. Biochim Biophys Acta 1094, 51–54 (1991).
Millart, H., Durlach, V. & Durlach, J. Red blood cell magnesium concentrations: analytical problems and significance. Magnes Res 8, 65–76 (1995).
Kaitsuka, T. et al. Inactivation of TRPM7 kinase activity does not impair its channel function in mice. Sci Rep 4, 5718 (2014).
Clark, K. et al. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS One 3, e1876 (2008).
Ryazanova, L. V., Dorovkov, M. V., Ansari, A. & Ryazanov, A. G. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem 279, 3708–3716 (2004).
Ye, Q., Crawley, S. W., Yang, Y., Cote, G. P. & Jia, Z. Crystal structure of the alpha-kinase domain of Dictyostelium myosin heavy chain kinase A. Sci Signal 3, ra17 (2010).
Touyz, R. M. Magnesium in clinical medicine. Front Biosci 9, 1278–1293 (2004).
Malpuech-Brugere, C. et al. Accelerated thymus involution in magnesium-deficient rats is related to enhanced apoptosis and sensitivity to oxidative stress. Br J Nutr 81, 405–411 (1999).
Rayssiguier, Y., Gueux, E., Bussiere, L., Durlach, J. & Mazur, A. Dietary magnesium affects susceptibility of lipoproteins and tissues to peroxidation in rats. J Am Coll Nutr 12, 133–137 (1993).
Rock, E. et al. Dietary magnesium deficiency in rats enhances free radical production in skeletal muscle. J Nutr 125, 1205–1210 (1995).
Weglicki, W. B. et al. Role of free radicals and substance P in magnesium deficiency. Cardiovasc Res 31, 677–682 (1996).
Weglicki, W. B. et al. Cytokines, neuropeptides and reperfusion injury during magnesium deficiency. Ann N Y Acad Sci 723, 246–257 (1994).
Aarts, M. et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877 (2003).
Sun, H. S. et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci 12, 1300–1307 (2009).
Rodushkin, I. & Axelsson, M. D. Application of double focusing sector field ICP-MS for multielemental characterization of human hair and nails. Part I. Analytical methodology. Sci Total Environ 250, 83–100 (2000).
Acknowledgements
This study was supported by NIH Grant P01GM078195 (A.G.R., A.F., V.C.). V.C. was also supported by the Deutsche Forschungsgemeinschaft (grants CH1181/1-1; TRP 152/1). We would like to thank Nikolay Kornakov and Vadim Kotov for the help in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.V.R. performed experiments, analyzed data and wrote the manuscript. Z.H. and S.S. performed experiments and V.C. and A.F. interpreted and analyzed data and edited the manuscript and A.G.R. conceived the study, analyzed data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ryazanova, L., Hu, Z., Suzuki, S. et al. Elucidating the role of the TRPM7 alpha-kinase: TRPM7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci Rep 4, 7599 (2014). https://doi.org/10.1038/srep07599
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07599
This article is cited by
-
TRPM channels in health and disease
Nature Reviews Nephrology (2024)
-
TRPM7 kinase mediates hypomagnesemia-induced seizure-related death
Scientific Reports (2023)
-
Structural mechanism of TRPM7 channel regulation by intracellular magnesium
Cellular and Molecular Life Sciences (2022)
-
TRPM7 restrains plasmin activity and promotes transforming growth factor-β1 signaling in primary human lung fibroblasts
Archives of Toxicology (2022)
-
It takes more than two to tango: mechanosignaling of the endothelial surface
Pflügers Archiv - European Journal of Physiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
