
Abstract
The cholesterol reducing drugs, statins, exhibit anti-tumor effects against cancer stem cells and various cancer cell lines, exert potent additivity or synergy with existing chemotherapeutics in animal models of cancer and may reduce cancer incidence and cancer related mortality in humans. However, not all tumor cell lines are sensitive to statins and clinical trials have demonstrated mixed outcomes regarding statins as anticancer agents. Here, we show that statin-induced reduction in intracellular cholesterol levels correlate with the growth inhibition of cancer cell lines upon statin treatment. Moreover, statin sensitivity segregates with abundant cytosolic vimentin expression and absent cell surface E-cadherin expression, a pattern characteristic of mesenchymal-like cells. Exogenous expression of cell surface E-cadherin converts statin- sensitive cells to a partially resistant state implying that statin resistance is in part dependent on the tumor cells attaining an epithelial phenotype. As metastasizing tumor cells undergo epithelial to mesenchymal transition during the initiation of the metastatic cascade, statin therapy may represent an effective approach to targeting the cells most likely to disseminate.
Similar content being viewed by others


Introduction
Repurposing existing drugs for new clinical applications is one of the safest and least resource-intensive approaches to improve therapeutic options1,2. In this regard, the cholesterol lowering drugs, statins, have been reported to reduce cancer incidence and cancer related mortality in patients3,4. Similarly, many in vitro experiments have shown antitumor effects of statins against cancer stem cells5,6 and various cancer cell lines through suppression of cell proliferation and/or induction of apoptosis7,8,9. Statins also exert potent additivity or synergy with existing chemotherapeutics. For example, fluvastatin combined with trastuzumab (a monoclonal antibody against ErbB2) provides potent synergistic cytotoxic effects in human breast cancer cell lines10. Moreover, fluvastatin or simvastatin significantly inhibited mammary tumor growth in ErbB2-transformed Neu transgenic mice11. However, not all tumor cell lines are sensitive to statins and clinical trials have reported mixed outcomes regarding statins as anticancer agents7,8,9.
Metabolic reprogramming is inherent to tumor growth and transformed cells require increased energy and metabolic precursors to build the tumor cell biomass12,13. In addition, the metabolite-induced alteration of epigenetic and regulatory states is also integral to tumor progression14,15. Metabolic alteration of cholesterol synthesis is one pathway that is linked to tumorigenesis and some cancer stem cells and cell lines exhibit increased cholesterol synthesis through the mevalonate pathway5,16. Statins exert their antitumor effect through their interference with tumor metabolism by inhibiting the enzyme, HMG-CoA reductase (HMGCR) that catalyzes the rate limiting step of the mevalonate/cholesterol synthesis pathway7,8,9 (Supplementary Fig. S1). Statin inhibition of HMGCR decreases the levels of mevalonate and its downstream products, including cholesterol, dolichol, ubiquinone and the isoprenoid intermediates geranyl-geranyl pyrophosphate and farnesyl pyrophosphate (Supplementary Fig. S1).
The metabolic state of tumor cells, however, is not uniform. Cancer cell lines range from small, highly proliferative cells to large, slowly proliferating mesenchymal-like cells and the growth inhibitory activity of statins is more potent against the latter type17. Yet, biomarkers that demarcate statin sensitive cancer cell lines have not been truly discerned, hampering their rational development as an adjuvant therapy.
Here, we show that statin-sensitive cancer cell lines exhibit mesenchymal-like phenotypes, characterized by abundant cytosolic vimentin and absent cell surface E-cadherin expression. In the presence of atorvastatin, these cell lines deplete their cholesterol, an effect that is circumvented by the simultaneous addition of mevalonate to the cell culture. Moreover, exogenous expression of cell surface E-cadherin converts statin-sensitive cells to a partially resistant state implying that statin resistance is in part dependent on intact E-cadherin signaling. As metastasizing tumor cells undergo epithelial to mesenchymal transition (EMT) during the initiation of the metastatic cascade from the primary tumor site18, statin co-therapy may be an effective approach to reduce the metastatic competency of primary tumors and the rate of metastasis formation.
Results
Variable growth inhibition of cancer cell lines in response to atorvastatin treatment
Previous experiments have demonstrated that statins, including atorvastatin (Lipitor), inhibit the growth of a subset of the NCI-60 cancer cell lines and if one statin inhibited the proliferation of a given cell line, then the other statins also showed similar half maximal inhibitory concentration (IC50) values19. To confirm these results, we cultured two cell lines from each of seven organ types obtained from the NCI-60 collection in standard growth medium with 10 µM atorvastatin. We found that atorvastatin affected the proliferation rates of these cancer cell lines differentially: the proliferation of some cell lines were fully or partially inhibited by atorvastatin while others were insensitive to it (Fig. 1). The growth inhibition in these cell lines does not correlate with increased levels of select apoptosis markers (data not shown), implying that statin treatment induces growth arrest.

Growth rate of atorvastatin treated NCI-60 cancer cell lines.
Colon cancer (A. HCT-116 and B. KM-12), ovarian cancer (C. IGROV1 and D. OVCAR3), breast cancer (E. HS-578T and F. T47D), lung cancer (G. HOP-92 and H. NCI-H322M), prostate cancer (I. PC-3 and J. DU-145), melanoma (K. SK-MEL-5 and L. MDA-MB-435) and brain cancer (M. SF-295 and N. SF-539) cell lines from the NCI-60 cancer cell line collection were treated with 10 µM atorvastatin (purple line) or DMSO vehicle control (blue line) and cell proliferation was quantified at 2, 4 and 6 days by direct cell counting. Each value represents the mean ± standard deviation (SD) (n = 3 for each group). The data were analyzed using Student's t-test. * P<0.05, ** P<0.01.
The addition of mevalonate, the product of the HMGCR-catalyzed reaction (Supplementary Fig. S1), to the growth medium circumvents the effects of HMGCR inhibition or siRNA downregulation of HMGCR expression7, presumably by substituting for endogenous mevalonate. To examine whether atorvastatin exerts its growth inhibitory effect through HMGCR inhibition, we added mevalonate to atorvastatin-treated cells. We found that mevalonate reversed the atorvastatin-induced growth inhibition of the ten cancer cell lines that displayed full or partial sensitivity to atorvastatin (Fig. 1; Supplementary Fig. S2) in a dose-dependent manner (Supplementary Fig. S3). These cell lines express HMGCR at comparable levels (Supplementary Fig. S4), demonstrating that statin sensitivity is not due to differential enzyme expression.
Atorvastatin sensitivity correlates with decreased cholesterol levels in atorvastatin-treated cells
Atorvastatin inhibits the enzymatic activity of HMGCR, thus blocking the synthesis of mevalonate and its downstream products that include cholesterol (Supplementary Fig. S1). To directly test the relationship between atorvastatin sensitivity and cholesterol levels, we measured the cholesterol content of the cell lines in the presence or absence of atorvastatin using Filipin III, a pentaene macrolide routinely used to assess intracellular cholesterol levels20. Representative results are shown in Figure 2 (the entire data set is shown in Supplementary Fig. S5A–C). Atorvastatin treatment drastically reduced the levels of intracellular cholesterol in atorvastatin sensitive cell lines when compared to that of the control DMSO treatment (Fig. 2G–I). In contrast, addition of the drug did not affect the levels of intracellular cholesterol in atorvastatin resistant cell lines (Fig. 2A–C). Cell lines with partial atorvastatin sensitivity displayed an intermediate phenotype upon atorvastatin treatment (Fig. 2D–F). Thus, the sensitivity of cell lines to atorvastatin-induced growth inhibition inversely correlated with the intracellular cholesterol levels following atorvastatin treatment.

Cholesterol content of atorvastatin treated NCI-60 cancer cell lines.
Cell lines were treated with 10 µM atorvastatin or 0.1% DMSO for 24 hours. Intracellular cholesterol levels were determined by Filipin III staining. Results for (A–C) atorvastatin resistant (DU-145), (D–F) partially sensitive (T47D) and (G–I) sensitive cell lines (HOP-92) are shown. The full data set for all fourteen NCI-60 cell lines is shown in Supplementary Figure S5. (A, D, G) DMSO control; (B, E, H) 10 μM atorvastatin treated cells; (C, F, I) histogram of cholesterol levels normalized to the DMSO controls. Each value represents the mean ± SD (n = 3 for each group). The data were analyzed using Student's t-test. ** P<0.01.
To further confirm that atorvastatin reduces the cells' cholesterol level through HMGCR inhibition, we measured the cholesterol content in the two most atorvastatin-sensitive cell lines (HOP-92, PC-3) 24 hours after the atorvastatin treatment in the presence or absence of mevalonate supplementation. As expected, we found that the addition of mevalonate to the growth medium prevented the atorvastatin-induced depletion of cholesterol typically seen in atorvastatin sensitive cell lines (Supplementary Fig. S6).
Total vimentin and E-cadherin expression are not suitable markers for statin sensitivity
The differential sensitivity of tumor cell lines to atorvastatin could be utilized for adjuvant cancer therapy if highly sensitive biomarkers were identified. Our previous analysis of the NCI-60 cancer cell lines indicated that cells with larger diameters in culture are more sensitive to statin treatment. Furthermore, increased cell size in culture correlated with higher vimentin protein expression, a standard marker of mesenchymal cell lineage, while the epithelial marker E-cadherin negatively correlated with increased cell size17. As these two markers are routinely used in standard immunohistochemistry of tumor biopsy specimens, we probed their utility as biomarkers of atorvastatin sensitivity.
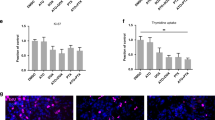
First, we performed Western blot analyses to determine the average vimentin and E-cadherin expression in the fourteen cell lines. We found that several cell lines express vimentin with absent or minimal E-cadherin expression while cell lines deficient in vimentin expressed a medium to high level of E-cadherin (Fig. 3). Of note, some cell lines expressed both vimentin and E-cadherin (Fig. 3); however, they also exhibited highly variable atorvastatin sensitivity. For example, the PC-3 and DU-145 prostate cancer cell lines express both proteins at comparable levels (Fig. 3), yet their responses to atorvastatin are completely opposite (Fig. 1). These data indicate that the correlation between mesenchymal marker vimentin and epithelial marker E-cadherin expression cannot be used as dependable biomarker for predicting or identifying atorvastatin sensitive cancer cells.

E-cadherin and vimentin expression of NCI-60 cancer cell lines.
The expression levels of E-cadherin and vimentin in the indicated fourteen NCI-60 cell lines were determined by western blotting. β-actin expression was used as the loading control. Representative images from three experiments are shown.
Cytoplasmic vimentin expression without concomitant membrane E-cadherin expression is a potential biomarker for atorvastatin sensitivity
While vimentin is a cytoplasmic protein, E-cadherin can be found in different subcellular localizations within tumor cells21,22. E-cadherin homodimerization at the cell membrane provides survival signals whereas destabilized E-cadherin are internalized and fails to signal23. To uncover the potential differences in E-cadherin localization in the fourteen cell lines, we performed double immunostaining for vimentin and E-cadherin expression and imaged using immunofluorescence microscopy.
Based on the collected data, the fourteen cell lines were segregated into one of three groups, each with different patterns of vimentin and E-cadherin expression (Fig. 4). First, six cell lines without cell surface E-cadherin expression but uniformly expressed cytoplasmic vimentin were atorvastatin-sensitive (Fig. 4A–F). These sensitive cell lines differed in their total E-cadherin content: HOP-92 and SK-MEL-5 cells showed discernible intracellular E-cadherin expression, whereas E-cadherin expression was undetectable in the other four cell lines (Supplementary Fig. S7).

E-cadherin and vimentin expression and subcellular localization.
Merged images of the NCI-60 cell lines immunostained for E-cadherin (green, membrane, cytoplasmic or nuclear), vimentin (red, cytoplasmic) and Hoechst 33342 (blue, nucleus). A: PC-3, B: HOP-92, C: SK-MEL-5, D: MDA-MB-435, E: SF-295, F: SF-539, G: HS-578T, H: T47D, I: HCT-116, J: IGROV1, K: DU-145, L: NCI-H322M, M: OVCAR3 and N: KM-12 cell lines. Cell lines are labeled as atorvastatin-sensitive (A–F), -partially sensitive (G–J) or -resistant (K–N) based on the results of cell proliferation studies shown in Figure 1.
In contrast, cell lines that displayed any cell membrane E-cadherin expression were either fully or partially atorvastatin-resistant (Fig. 4H–N), irrespective of their cytoplasmic vimentin expression. The degree of vimentin and E-cadherin expression varied among the individual cell lines. For example, DU-145 cells display both cytoplasmic vimentin and heterogeneous cell membrane E-cadherin expression (Fig. 4K, Supplementary Fig. S8), whereas OVCAR3 cells are vimentin negative but exhibit both cell membrane and intracellular E-cadherin localization (Fig. 4M, Supplementary Fig. S8). Despite this difference, both cell lines are fully resistant to atorvastatin (Fig. 1).
A similar variation exists among partially resistant cell lines. T47D and HCT-116 cells are positive for cell membrane and cytoplasmic E-cadherin but are vimentin negative (Fig. 4H, I), whereas IGROV1 cell line is comprised of at least two different cell types. A subset of IGROV1 cells are vimentin negative and positive for cell membrane E-cadherin while another subset of cells is positive for cytoplasmic vimentin and exhibit cell membrane and cytoplasmic E-cadherin (Fig. 4J, Supplementary Fig. S9).The only exception is the HS-578T breast cancer cell line that is only partially atorvastatin sensitive (Fig. 1), even though it has similar vimentin and E-cadherin expression pattern as that observed in atorvastatin-sensitive cell lines (Fig. 4G, Supplementary Fig. S9).
These results suggest that cell membrane E-cadherin expression is a marker of full or partial atorvastatin resistance, whereas uniform vimentin expression without any cell membrane E-cadherin expression largely correlates with atorvastatin sensitivity.
Forced expression of cell surface E-cadherin converts atorvastatin-sensitive cells to a partially atorvastatin resistant state
Given the observed correlation between atorvastatin-resistance and cell membrane E-cadherin expression, we aimed to determine whether exogenously expressed membrane E-cadherin would induce a resistant state in a statin-sensitive cell line. To this end, we used derivatives of a highly statin-sensitive MDA-MB-231 cell line, which express either dsRED (a RFP variant) alone or dsRED and cell surface E-cadherin (Ecad)24. The RFP and Ecad cell lines were treated with log-dilutions of atorvastatin and their cell numbers were determined 3 days after treatment by crystal violet staining. We found that the half maximal inhibitory concentration of atorvastatin (IC50) in MDA-MB-231 cells shifted from 1.16 μM to 4.3 μM when cell surface E-cadherin was expressed (Fig. 5A). A representative image of RFP and Ecad cells treated with 3 µM atorvastatin illustrates this shift (Fig. 5B,C). In contrast, a PC-3 cell line with high exogenous cytoplasmic E-cadherin expression did not show the same development of resistance to atorvastatin (Supplementary Fig. S10). These data suggest that forced cell surface E-cadherin expression can confer partial statin resistance to formerly statin-sensitive cancer cells.

Effect of forced cell surface E-cadherin expression on atorvastatin sensitivity.
(A) MDA-MB-231 RFP ( ) or MDA-MB-231 RFP E-cadherin (
) or MDA-MB-231 RFP E-cadherin ( ) cells were treated with 0.1 µM, 0.3 µM, 1 µM, 3 µM, 10 µM, or 30 µM atorvastatin. Cell viability was assessed by crystal violet staining 3 days after atorvastatin treatment and normalized to the 0.3% DMSO control. The data were fit with a sigmoid function to extrapolate the IC50 value (lines). Each value represents the mean ± SD (n = 3 for each group). The data for each MDA-MB-231 RFP E-cadherin cells were compared with those for the controls (MDA-MB-231 RFP cells). Asterisk, two-way ANOVA (P<0.01) followed by t-test (P<0.05). (B) MDA-MB-231 RFP and (C) MDA-MB-231 RFP E-cadherin cells were treated with 3 µM atorvastatin. Imaging was done with 40X oil objective lens 3 days after the treatment.
) cells were treated with 0.1 µM, 0.3 µM, 1 µM, 3 µM, 10 µM, or 30 µM atorvastatin. Cell viability was assessed by crystal violet staining 3 days after atorvastatin treatment and normalized to the 0.3% DMSO control. The data were fit with a sigmoid function to extrapolate the IC50 value (lines). Each value represents the mean ± SD (n = 3 for each group). The data for each MDA-MB-231 RFP E-cadherin cells were compared with those for the controls (MDA-MB-231 RFP cells). Asterisk, two-way ANOVA (P<0.01) followed by t-test (P<0.05). (B) MDA-MB-231 RFP and (C) MDA-MB-231 RFP E-cadherin cells were treated with 3 µM atorvastatin. Imaging was done with 40X oil objective lens 3 days after the treatment.
Discussion
Tissue dysfunction and cachexia due to growing metastases is the cause of death in most cancer patients. Conceptually, the formation of cancer metastases follows a cascade of events18. Initially, a fraction of cancer cells in the primary tumor undergo EMT18,25 allowing them to detach and intravasate into the circulation. These circulating, mesenchymal-like tumor cells then reach distant organs whose successful colonization requires a subsequent extravasation and partial reversion of the tumor cells' phenotype by mesenchymal to epithelial reverting transition (MErT)18,26. After colonization, a variable length of dormancy ensues27,28 followed by outgrowth of these dormant micrometastases that requires at least a partial return to EMT18. The challenge for the treatment of clinically evident metastases and silent micrometastases (which are likely asynchronous in individual metastatic nodules) is that existing chemotherapeutic strategies against them are not effective. Therefore, there is a dire need for pharmacotherapy that can reduce the metastatic competency of primary tumors by targeting primary tumor cells that undergo EMT and adopt a mesenchymal-like state.
In this study, we show that statins are potential drugs for such a therapeutic purpose. We demonstrate that atorvastatin sensitive cancer cell lines are mesenchymal-like with abundant cytosolic vimentin and no cell surface E-cadherin expression (Fig. 4A–F). In the presence of atorvastatin, the cholesterol levels of these sensitive cell lines were severely reduced and this effect was circumvented by the simultaneous addition of mevalonate to the cell culture medium. While abundant cytosolic vimentin and absent cell surface E-cadherin expression appear to be apt biomarkers for determining atorvastatin sensitivity, the HS-578T breast cancer cell line that display such a profile (Fig. 4G) is only partially resistant to atorvastatin (Fig. 1E). Excluding the HS-578T cell line, all partially or fully atorvastatin resistant cell lines express some degree of cell surface E-cadherin (Fig. 4H–N). Moreover, cell surface E-cadherin expression mediates the statin resistance to some degree as observed by the partial statin resistance that was induced by exogenous membrane E-cadherin expression in the mesenchymal MDA-MB-231 cell line (Fig. 5) while cytoplasmic E-cadherin expression in the mesenchymal PC-3 cell line (Supplementary Fig. S10) was without effect. Other aspects contributing to statin resistance may include differences in gene expression, post-translational modifications, compensatory upregulation of HMGCR after statin therapy9, or enhanced export of statin from the cells.
In statin-sensitive cell lines, the inhibition of HMGCR likely leads to growth inhibition by at least three different mechanisms. First, a mevalonate pathway-produced metabolite, geranyl-geranyl pyrophosphate inhibits Rho GTPases, which activate transcription factors YAP and TAZ of the Hippo pathway29,30,31 (Supplementary Fig. S1). These transcription factors are not only crucial for normal organ size control and stem cell renewal but also play an important role in inducing tumorigenesis and metastasis29,32, in part by inducing mesenchymal differentiation33, cancer stem cell related traits34 and cancer cell motility30. Therefore, statin inhibition of HMGCR may arrest cancer cell proliferation by inactivating downstream transcription factors. Second, statins impair the glucose uptake of tumor cells35. This effect is potentially related to the decreased cholesterol concentration in the cell membrane, which is crucial for membrane lipid raft functions that govern the subcellular localization and function of glucose transporters35 and other receptor complexes36. Third, accumulation of metabolic precursors, such as acetyl CoA, could also block glucose uptake through feedback inhibition of the glycolysis pathway37.
Future studies will aim at examining the vimentin and E-cadherin expression and functional state of circulating tumors cells. Furthermore, the susceptibility of these tumor cells to statin treatment will be explored using both in vitro cell cultures and an all-human microphysiological system that can recapitulate the early and later stages of micrometastasis formation38. The molecular mode of statin-sensitivity will also be discerned to determine if the key targets are prenylation as opposed to cholesterol deficiency. Finally, statin-drug combination therapies targeting downstream molecules of the mevalonate pathway will be tested as a means to potentiate the sensitivity of partially sensitive mesenchymal-like tumor cells and to reduce the therapeutic dose of statins to a suitable level for routine clinical use in oncology.
Methods
Cell cultures
We have selected 7 pairs of cell lines from the NCI-60 cancer cell panel representing 7 different major solid tumor types. For each site we selected a cell line with low and one with high protein synthesis rate, as previously reported17. (The selection was performed prior to the vimentin and E-cadherin profiling for mesenchymal and epithelial phenotype). The selected cell lines—colon cancer (HCT-116 and KM-12), ovarian cancer (IGROV1 and OVCAR3), breast cancer (HS-578T and T47D), lung cancer (HOP-92 and NCI-H322M), prostate cancer (PC-3 and DU-145), melanoma (SK-MEL-5 and MDA-MB-435) and brain cancer (SF-295 and SF-539)—were cultured in RPMI 1640 medium (Life Technologies, Grand Island, NY), supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS, Life Technologies) and 1% penicillin/streptomycin (Life Technologies) at 37°C with 5% CO2.
Atorvastatin treatment and cell proliferation assay
Atorvastatin (Sigma-Aldrich, St. Louis, MO) at a final concentration of 10 µM was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich; final concentration of 0.1% in RPMI 1640 medium). The cells were seeded in 6-well plates at a density of 1 × 105 cells/ml and incubated overnight prior to treating with 10 µM atorvastatin or 0.1% DMSO, which served as a control. Three independent experiments were performed. Cell proliferation was quantified at 2, 4 and 6 days by direct cell counting with Scepter™ Handheld Automated Cell Counter (EMD Millipore, Billerica, MA) using Scepter™ Tips-60 µm sensor (EMD Millipore), or by crystal violet staining. Preliminary studies demonstrated that the two methods yielded congruent results.
Mevalonic acid treatment in atorvastatin sensitive cells
To determine whether mevalonic acid treatment reverted the atorvastatin-sensitive phenotype, the cell lines whose proliferation was inhibited by atorvastatin were seeded in 6-well plates at a density of 1 × 105 cells/ml, incubated overnight and then treated with 10 µM atorvastatin and various concentrations (25 µM, 50 µM, 100 µM and 200 µM) of R-mevalonic acid (Sigma-Aldrich) for 24 hours. These cells were also photographed with a phase-contrast microscope to capture any morphological changes.
Cholesterol staining
Subconfluent (40–60% confluency) cells grown on coverslips in a 24-well plate were incubated with RPMI 1640 medium supplemented with 10% HI-FBS and 10 µM atorvastatin. To increase the visibility of the synthesized cholesterols, a cholesterol intercellular trafficking inhibitor, U18666A (Cayman Chemical, Ann Arbor, MI) was added to the culture medium at a concentration of 1.25 µM. Some of the atorvastatin sensitive cells (HOP-92 and PC-3) were also treated with 100 µM R-mevalonic acid (Sigma-Aldrich). Cells treated with 0.1% DMSO and 1.25 µM U18666A served as control. After 24-hours incubation, the cells were stained using the cholesterol cell-based detection assay kit (Cayman Chemical) according to the manufacturer's instructions. Briefly, the cells were fixed with 4% formaldehyde in Tris-buffered saline (TBS) for 10 min and then washed in TBS with 0.1% Tween 20 (TBS-T). The cells were incubated with 50 μg/ml Filipin III in TBS, a probe used for the fluorescent detection of sterols (excitation/emission, 340–380/385–470 nm). After 1 hour incubation with Filipin III in the dark at room temperature (RT), the cells were washed in TBS-T, followed by detachment and mounting of the coverslips in the aqueous-based mounting medium (Clearmount™, Invitrogen, Carlsbad, CA). Images were taken under an Olympus Provis fluorescence microscope (Olympus Optical, Tokyo, Japan) equipped with a 40X oil objective lens. Quantification of the cholesterol levels was accomplished using the Granularity algorithm in the MetaXpress image analysis software (Molecular Devices Corp., Sunnyvale CA). The total integrated granule intensity was measured for each image and the percent cholesterol remaining after atorvastatin treatment was calculated as the ratio of granule intensity in the atorvastatin cells to that of the DMSO treated cells.
Western blotting
After removing spent media and washing cells with cold phosphate buffered saline (PBS), the cells were incubated with cold Pierce RIPA lysis buffer (Thermo Scientific, Hudson, NH) containing Halt™ Protease Inhibitor Single-Use Cocktail, Halt™ Phosphatase Inhibitor Single-Use Cocktail (Thermo Scientific) and 1 mM dithiothreitol (DTT) for 15 min with occasional swirling. Then, the cells were scraped, homogenized with a 26-gauge needle and vortexed at the highest setting for 1 min; the lysates were cleared by centrifuging at 16,000 g at 4°C for 15 min. Protein concentration was determined with the bicinchoninic acid (BCA) method (BCA Protein Assay - Reducing Agent Compatible; Thermo Scientific). Proteins were separated on NuPAGE 4–12% Bis Tris gel electrophoresis (Life Technologies) and transferred to a nitrocellulose membrane (iBlot Gel Transfer Stacks Nitrocellulose; Life Technologies) using iBlot Gel Transfer Device (Life Technologies). The membrane was probed with monoclonal rabbit antibodies, anti-HMGCR (1:500, ab174830, Abcam Inc., Cambridge, MA), anti-vimentin (1:1000, ab92547, Abcam) and anti-E-cadherin (1:1000, 24E10, Cell Signaling Technology, Beverly, MA). A monoclonal mouse antibody to β-actin (1:500, ab8226, Abcam) was used as a loading control. Immunodetection was performed using the iBlot Western Detection chromogenic kit (Life Technologies).
Immunofluorescence microscopy
Cultured cells grown on coverslips in a 24-well plate were fixed with 2% paraformaldehyde (Sigma-Aldrich) for 30 min, washed in PBS and then permeabilized with 0.1% Triton-X-100 (Fisher Scientific, Pittsburgh, PA) made in PBS for 15 min. Following a PBS wash, non-specific proteins were blocked in 2% BSA for 15 min at RT. The cells were incubated with a mixture of two primary antibodies: monoclonal rabbit antibody to vimentin (1:100, ab92547, Abcam) and monoclonal mouse antibody to E-cadherin (1:50, ab1416, Abcam) in a humidified atmosphere for 1 hour at 37°C. Coverslips were then probed with Alexa Fluor 488 goat anti-mouse IgG and Alexa Fluor 555 goat anti-rabbit IgG (both; 1:200, Abcam) in the dark for 15 min at RT. Following a PBS wash, nuclei were stained with Hoechst 33342 (50 μg/ml) for 5 min at RT, washed and mounted in an aqueous-based mounting medium Clearmount™ (Invitrogen). Images were captured with the 40X oil objective lens on the Olympus Provis fluorescence microscope (Olympus Optical).
IC50 determination
MDA-MB-231 (breast cancer) and PC-3 (prostate cancer) cell lines were seeded in 12-well plates at a concentration of 1 × 105 cells/ml. The next day, cells were treated with 0.1 µM, 0.3 µM, 1 µM, 3 µM, 10 µM, or 30 µM atorvastatin. Cells treated with 0.3% DMSO (the concentration of DMSO in the 30 µM atorvastatin treatment condition) served as a control group. Three days after treatment, the cells were washed with PBS and fixed in 4% formaldehyde (F79-1, Fisher Scientific) for 15 minutes. The cells were washed with milli-Q water and stained with 0.5% w/v crystal violet (Sigma-Aldrich) for 10 minutes and excess dye was washed extensively with tap water. The absorbed dye was released with 2% SDS. The supernatants were mixed thoroughly before transferring to a 96-well plate to be read at 560 nm using a Tecan SpectraFluor microplate reader (Tecan US, Durham, NC). IC50 values were determined by fitting a standard, four-parameter sigmoid curve to the data. All treatments were carried out in triplicate samples.
Statistical analyses
Statistical analyses were performed using the StatView software (version 5.0; SAS Institute Inc., Cary, NC). The data for atorvastatin-treated cells and non-treated controls were compared using two-way analysis of variance (ANOVA) and Student's t-test. P-values less than 0.05 were regarded statistically significant.
References
Issa, N. T., Byers, S. W. & Dakshanamurthy, S. Drug repurposing: translational pharmacology, chemistry, computers and the clinic. Curr. Top. Med. Chem. 13, 2328–2336 (2013).
Stenvang, J. et al. Biomarker-guided repurposing of chemotherapeutic drugs for cancer therapy: a novel strategy in drug development. Front. Oncol. 3, 313, 10.3389/fonc.2013.00313 (2013).
Farwell, W. R. et al. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 100, 134–139, 10.1093/jnci/djm286 (2008).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 367, 1792–1802, 10.1056/NEJMoa1201735 (2012).
Ginestier, C. et al. Mevalonate metabolism regulates basal breast cancer stem cells and is a potential therapeutic target. Stem Cell 30, 1327–1337, 10.1002/stem.1122 (2012).
Hartwell, K. A. et al. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat. Chem. Biol. 9, 840–848, 10.1038/nchembio.1367 (2013).
Gazzerro, P. et al. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol. Rev. 64, 102–146, 10.1124/pr.111.004994 (2012).
Osmak, M. Statins and cancer: current and future prospects. Cancer Lett. 324, 1–12, 10.1016/j.canlet.2012.04.011 (2012).
Clendening, J. W. & Penn, L. Z. Targeting tumor cell metabolism with statins. Oncogene 31, 4967–4978, 10.1038/onc.2012.6 (2012).
Budman, D. R., Tai, J. & Calabro, A. Fluvastatin enhancement of trastuzumab and classical cytotoxic agents in defined breast cancer cell lines in vitro. Breast Cancer Res. Treat. 104, 93–101, 10.1007/s10549-006-9395-5 (2007).
Campbell, M. J. et al. Breast cancer growth prevention by statins. Cancer Res. 66, 8707–8714, 10.1158/0008-5472.can-05-4061 (2006).
Levine, A. J. & Puzio-Kuter, A. M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344, 10.1126/science.1193494 (2010).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033, 10.1126/science.1160809 (2009).
Li, F., Xu, W. & Zhao, S. Regulatory roles of metabolites in cell signaling networks. J. Genet. Genomics 40, 367–374, 10.1016/j.jgg.2013.05.002 (2013).
Venneti, S. & Thompson, C. B. Metabolic modulation of epigenetics in gliomas. Brain Pathol. 23, 217–221, 10.1111/bpa.12022 (2013).
Li, Y. C., Park, M. J., Ye, S. K., Kim, C. W. & Kim, Y. N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 168, 1107–1118; 10.2353/ajpath.2006.050959 (2006).
Dolfi, S. C. et al. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 1, 20, 10.1186/2049-3002-1-20 (2013).
Chaffer, C. L. & Weinberg, R. A. A perspective on cancer cell metastasis. Science 331, 1559–1564, 10.1126/science.1203543 (2011).
Shoemaker, R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823, 10.1038/nrc1951 (2006).
Ginsbach, C. & Fahimi, H. D. Labeling of cholesterol with filipin in cellular membranes of parenchymatous organs. Standardization of incubation conditions. Histochemistry 86, 241–248 (1987).
Chetty, R., Serra, S. & Asa, S. L. Loss of membrane localization and aberrant nuclear E-cadherin expression correlates with invasion in pancreatic endocrine tumors. Am. J. Surg. Pathol. 32, 413–419, 10.1097/PAS.0b013e31813547f8 (2008).
Han, A. C., Soler, A. P., Tang, C. K., Knudsen, K. A. & Salazar, H. Nuclear localization of E-cadherin expression in Merkel cell carcinoma. Arch. Pathol. Lab. Med. 124, 1147–1151, 10.1043/0003-9985 (2000).
Chao, Y., Wu, Q., Shepard, C. & Wells, A. Hepatocyte induced re-expression of E-cadherin in breast and prostate cancer cells increases chemoresistance. Clin. Exp. Metastasis 29, 39–50, 10.1007/s10585-011-9427-3 (2012).
Chao, Y. L., Shepard, C. R. & Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 9, 179, 10.1186/1476-4598-9-179 (2010).
Yu, M. et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584, 10.1126/science.1228522 (2013).
Chao, Y., Wu, Q., Acquafondata, M., Dhir, R. & Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 5, 19–28, 10.1007/s12307-011-0085-4 (2012).
Taylor, D. P., Wells, J. Z., Savol, A., Chennubhotla, C. & Wells, A. Modeling boundary conditions for balanced proliferation in metastatic latency. Clin. Cancer Res. 19, 1063–1070, 10.1158/1078-0432.ccr-12-3180 (2013).
Wells, A., Griffith, L., Wells, J. Z. & Taylor, D. P. The dormancy dilemma: quiescence versus balanced proliferation. Cancer Res. 73, 3811–3816, 10.1158/0008-5472.can-13-0356 (2013).
Zhao, B., Tumaneng, K. & Guan, K. L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13, 877–883, 10.1038/ncb2303 (2011).
Wang, Z. et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA. 111, E89–98, 10.1073/pnas.1319190110 (2014).
Sorrentino, G. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 16, 357–366, 10.1038/ncb2936 (2014).
Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505, 10.1016/j.devcel.2010.09.011 (2010).
Bhat, K. P. et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 25, 2594–2609, 10.1101/gad.176800.111 (2011).
Cordenonsi, M. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772, 10.1016/j.cell.2011.09.048 (2011).
Malenda, A. et al. Statins impair glucose uptake in tumor cells. Neoplasia 14, 311–323 (2012).
George, K. S. & Wu, S. Lipid raft: A floating island of death or survival. Toxicol. Appl. Pharmacol. 259, 311–319, 10.1016/j.taap.2012.01.007 (2012).
Jenkins, C. M., Yang, J., Sims, H. F. & Gross, R. W. Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: a mechanism integrating glycolytic flux with lipid metabolism. J. Biol. Chem. 286, 11937–11950, 10.1074/jbc.M110.203661 (2011).
Wheeler, S. E. et al. All-human microphysical model of metastasis therapy. Stem Cell Res. Ther. 4 Suppl 1S11, 10.1186/scrt372 (2013).
Acknowledgements
This research was in part supported by a VA Merit grant and an NCATS-funded Tissue Chip program (TR000496) to A.W. and a National Science Foundation grant to Z.N.O.
Author information
Authors and Affiliations
Contributions
K.W., T.W., C.B., A.V., A.W. and Z.N.O. designed the experiments. K.W., T.W. and C.B. performed the experiments. K.W., T.W., C.B., A.V., M.E.S., A.W. and Z.N.O. analyzed the data. K.W., C.B., A.V., A.W. and Z.N.O. wrote the manuscript. All authors have reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Warita, K., Warita, T., Beckwitt, C. et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep 4, 7593 (2014). https://doi.org/10.1038/srep07593
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07593
This article is cited by
-
Paradoxical effects of statins on endothelial and cancer cells: the impact of concentrations
Cancer Cell International (2023)
-
Post-diagnostic statin use and breast cancer-specific mortality: a population-based cohort study
Breast Cancer Research and Treatment (2023)
-
Raman spectroscopy biochemical characterisation of bladder cancer cisplatin resistance regulated by FDFT1: a review
Cellular & Molecular Biology Letters (2022)
-
Computational pharmacogenomic screen identifies drugs that potentiate the anti-breast cancer activity of statins
Nature Communications (2022)
-
A review of effects of atorvastatin in cancer therapy
Medical Oncology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
