
Abstract
Lactobacillus acidophilus is a Gram-positive lactic acid bacterium that has had widespread historical use in the dairy industry and more recently as a probiotic. Although L. acidophilus has been designated as safe for human consumption, increasing commercial regulation and clinical demands for probiotic validation has resulted in a need to understand its genetic diversity. By drawing on large, well-characterised collections of lactic acid bacteria, we examined L. acidophilus isolates spanning 92 years and including multiple strains in current commercial use. Analysis of the whole genome sequence data set (34 isolate genomes) demonstrated L. acidophilus was a low diversity, monophyletic species with commercial isolates essentially identical at the sequence level. Our results indicate that commercial use has domesticated L. acidophilus with genetically stable, invariant strains being consumed globally by the human population.
Similar content being viewed by others

Introduction
The lactic acid bacteria (LAB) are a group of Gram-positive bacteria united by their ability to produce lactic acid as a major end product of carbohydrate metabolism1 which has resulted in their artisanal and industrial use in dairy fermentations2. A history of safe consumption of lactobacilli is widely acknowledged, but more recently specific LAB strains associated with health benefits have been sold as probiotics, establishing a market sector in excess of $100 billion annually2,3. Most of the marketed strains belong to the genera Bifidobacterium and Lactobacillus. The lactobacilli are a highly heterogeneous taxonomic group, encompassing species with a wide range of genetic, biochemical and physiological properties4. The number of validly named Lactobacillus species has considerably increased in the last 10–15 years, with 201 species currently described5,6. The standards for the designation of new Lactobacillus taxa have also been recently updated, underlining the importance of applying the same rigorous standards to previously described and taxonomically assigned isolates6.
Lactobacillus acidophilus is added to commercial yoghurts and dairy formulations both for its flavour and for probiotic effect and is one of the most commonly selected Lactobacillus species for dietary use7,8,9. Taxonomically, L. acidophilus has undergone multiple revisions concurrent with changes and enhancements in techniques to investigate taxonomic relationships, from biochemical tests, through DNA sequence-based approaches, to comparisons utilising the whole genome sequence (WGS)10. The phenotypic and biochemical characteristics of individual L. acidophilus isolates show evidence of diversity11,12; however, recent genotypic analyses indicate less variation is present within the L. acidophilus genome. PCR fingerprinting demonstrated that five independent L. acidophilus isolates grouped as a single strain genotype by Randomly Amplified Polymorphic DNA (RAPD) analysis13. A seven-locus Multi Locus Sequence Typing (MLST) scheme revealed that the L. acidophilus isolates examined encompassed just two MLST allelic profiles, with distinct isolates differing by a single nucleotide14. Recent studies independently examining the genomic sequences of three (NCFM, ATCC 4796 and La-14)15 and five (CIP 76.13T, CIRM-BIA 442, CIRM-BIA 445, DSM 20242 and DSM 9126)16L. acidophilus genomes, have also shown remarkable levels of genetic identity.
The limited infraspecific level diversity of L. acidophilus warrants further investigation in light of its taxonomy, worldwide commercial use and food-based consumption and the increasing clinical role of probiotics. High throughput DNA sequencing enables rapid acquisition of bacterial WGS allowing researchers to comprehensively interrogate phylogenetic relationships between groups of bacterial isolates at taxonomic levels from domain to strain17. Here, we elucidate the infraspecific phylogenetic relationships between L. acidophilus isolates using reference-free, de novo assembly of whole genome sequence data, combined with hierarchical gene-by-gene analysis of whole genome sequences (WGS)18. Examination of a collection of L. acidophilus isolates spanning a timeframe of 92 years, multiple geographic locations and distinct sources, demonstrated an absence of genetic diversity within the species and WGS identity for strains in current commercial use.
Results
L. acidophilus lacks genomic diversity compared to other L. acidophilus group species
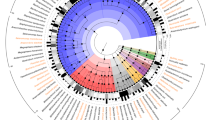
Within the bacterial genus Lactobacillus multiple phylogenetic groups have been defined based on analysis of 16S rRNA gene sequences, with the most recent analysis placing L. acidophilus within a cluster designated as the Lactobacillusdelbrueckii group19. The same group of closely related Lactobacillus species has also been referred to as the L. acidophilus group in multiple previous studies14,20,21 and this terminology is adopted herein. Despite the widespread use of L. acidophilus, the number of polyphasically identified isolates of this species deposited within well characterised collections of LAB was limited and previous analysis had shown very little genetic diversity within this species9,13,14,16; 24 isolates and 10 L. acidophilus genomes were collected as representative of the available diversity of this species (Table 1). To contextualise the genetic diversity of L. acidophilus isolates at the species level, ribosomal MLST (rMLST)22 was used to evaluate the genomic diversity present in species representative of the L. acidophilus group. Fifty-three genes that encode ribosomal proteins (rps) were identified in all 32 L. acidophilus genome sequences examined (Table 1) and among 108 reference genomes available from species within the L. acidophilus group19 (Table S1). The L. acidophilus group (represented by nine species) encompassed 99 rMLST types, with no two isolates for an individual species other than L. acidophilus, sharing a single allelic profile. A phylogenetic network of rMLST gene nucleotide sequence variation within the L. acidophilus group was constructed using the Neighbour Net algorithm (Figure 1). All species were resolved and rMLST gene sequence diversity was evident at the infraspecific level in all species except for L. acidophilus, Lactobacillus iners and Lactobacillus ultunensis (Figure 1). Of the 53 rps loci examined in the rMLST analysis, just 13 showed sequence variability in L. acidophilus isolates. In contrast, all other L. acidophilus group species showed within-species variation at 41 or greater rps loci (ranging from 41 in L. iners to 51 in L. helveticus) (Figure 1). While all 9 L. acidophilus group species demonstrated clear separation in the rMLST gene Neighbour Net, L. acidophilus, L. crispatus, L. ultunensis and L. helveticus were linked by a polytomy, suggesting they had all evolved from a common ancestor (Figure 1).

L. acidophilus lacks genomic diversity compared to other L. acidophilus group species.
A NeighborNet graph of 32 L. acidophilus (Table 1) and 108 L. acidophilus group (Table S1) genomes was generated using the sequence of 53 concatenated rMLST loci, providing a least-squares fit of 99.99%. The number of genomes from each species included in the network are given in brackets adjacent to the species name label. The scale bar indicates uncorrected P distance measured as number of nucleotide differences over 9338 parsimony informative nucleotides.
The infraspecific diversity of L. acidophilus using whole genome MLST (wgMLST)
Further analysis of L. acidophilus genome sequences was conducted using DNA sequence from all complete protein-coding regions to improve resolution and enable infraspecific genetic diversity to be mapped. Of 1,864 loci defined in the L. acidophilus NCFM genome sequence23, Genome Comparator analysis18 identified 1,815 (97.4%) complete loci present in all the L. acidophilus genomes examined (Table 1). Of these 1,815 core loci, 972 (53.6%) showed sequence variation in at least one isolate. A NeighborNet analysis18 of allelic variation within all shared loci encoded in the L. acidophilus NCFM genome sequence was able to resolve each isolate within the L. acidophilus cluster and demonstrated the presence of a notable, highly conserved, sub-group comprised of commercial isolates (Figure 2).

Whole genome MLST analysis of the infraspecies diversity of L. acidophilus.
A Neighbor Net plot was generated using wgMLST. The scale bar indicates distance measured in number of allelic differences over 1815 genomic loci conserved across all L. acidophilus genomes examined. Isolate numbers are coloured to represent their commercial (blue) or culture collection (green) history (Table 1), with other notable groups circled. High resolution analysis of the 14 commercial L. acidophilus genome sequences in the circled region of the neighbour net plot is provided in Supplementary Figure S1. The NFCM isolate genome sequenced as part of this study is indicated by an asterisk. The GenBank accession of the NCFM reference genome sequence is given brackets. Numbers given in superscript indicate CRISPR sequence types assigned in Supplementary Figure S2.
Genome sequences generated from commercially used L. acidophilus strains (Table 1) and isolates cultivated directly from current probiotic products13 formed a tight cluster centred on the widely used L. acidophilus NCFM (Figure 2, labelled in blue). Only one isolate in commercial use within a dairy product (strain CIRM-BIA 445) placed outside the commercial isolate cluster (Figure 2); the phylogenetic relationships between commercial L. acidophilus isolates are shown at higher resolution in Supplementtary Figure S1 and the variable loci within these genomes are listed in Supplementary Table S2. The published L. acidophilus NCFM genome sequence (accession number CP000033) was generated in 2005 using Sanger sequencing23. This reference genome formed a distinct arm deriving from the central commercial isolate node and separated from a duplicate isolate of L. acidophilus NCFM that was re-sequenced as a control for this study (Figure 2; see asterisk). When the two NCFM genome sequences were compared, all loci defined in the published reference sequence23 were present in re-sequenced genome. However, 89 loci showed sequence differences and 26 of these variable loci from the NCFM re-sequence were found to have identical sequence in all other L. acidophilus isolates. This suggests that errors in the original NCFM genome23, that were corrected by the massively parallel sequencing reads used for the duplicate NCFM isolate examined, were the most probable source of this variation.
Comparison of the total number of loci with variable sequence between the commercial and type strain cluster isolates provided a measure of the limited variation within the industrial isolates. Within the commercial isolate cluster, 118 loci were found to have variable sequence in at least one isolate. By comparison, isolates from the type strain cluster, representative of a single strain deposited in duplicate locations or under a different alias, such as L. acidophilus LMG 9433T and L. acidophilus LMG 13550T (Table 1), variation in at least one isolate was observed at 337 loci. Additional evidence of genetic conservation of the commercial isolates was also seen in 6 loci constituting the prophage remnant designated Potentially Autonomic Unit 3 (PAU3)23; these were not detected in any of the type strain cluster sequences, yet this region was fully intact in all commercial isolates.
The genome sequences generated from other L. acidophilus isolates taken from different culture collections (Table 1) were also diffuse in their placement in the wgMLST NeighborNet (Figure 2). No major sub-clusters other than the type strain cluster were apparent in the culture collection genomes examined (Figure 2). However, in analogous fashion to the type strain cluster, close placement of identical isolates sequenced in different studies using distinct sequencing and assembly technologies was seen. For example, L. acidophilus LMG 11470 (Illumina HiSeq2000 and Velvet assembly, this study) and ATCC 4796 (454-GS-FLX, Newbler assembly, Human Microbiome Project) (Figure 2, Table 1).
Conservation of the L. acidophilus genome and limited protein-coding locus variation
To map the conservation of the L. acidophilus genome, a pairwise comparison of selected isolate sequences was carried out against the L. acidophilus NCFM annotated reference genome23 (CP00033; Figure 3). This analysis corroborated the wgMLST data (Figure 1 and 2) and demonstrated that genetic variation in the core genome of L. acidophilus was primarily comprised of single nucleotide polymorphisms (Figure 3). Novel DNA that did not map the NCFM reference genome was not found in any of the L. acidophilus isolate sequence assemblies. Only limited evidence of genetic loss was detected in the pairwise analysis, with the absent genomic regions confined to genes encoding phage-related, mucus binding and sugar metabolism functions (Figure 3).

L. acidophilus genome sequences compared to L. acidophilus NCFM.
The black, innermost ring represents the published genome sequence of L. acidophilus NCFM (accession CP000033). Further concentric rings correspond to isolates according to the key. Co-localising isolates from Figure 2 that could be linked by alias (Table 1) are combined into a single ring. Ring presence indicates 98% sequence identity. Regions of interest are annotated.
Three prophage remnants, designated as Potentially Autonomic Units (PAU; PAU1, PAU2 and PAU3) within the L. acidophilus NCFM genome23 and a novel region of three consecutive loci (LBA0058 to LBA0060) with phage related functions, demonstrated variable presence among the L. acidophilus isolates. Differences in the distribution of the three PAU regions was evident when the isolate history, as commercial or culture collection derived (Table 1), was considered. The PAU1 locus was widely distributed across all isolates with exception to the commercial isolates CUL21 and C47 (Figure 3). The remaining PAU regions, 2 and 3, were intact for all commercial isolates. However, the culture collection isolates demonstrated variable presence of loci within PAU2, PAU3 and the phage-related LBA0058-60 region (Figure 3). A region corresponding to L. acidophilus LBA1019-LBA1020, encoding mucus binding proteins, was also absent from culture collection isolates L. acidophilus LMG 11469, LMG 11472, LAB 69 and ATCC 4796/LMG 11470. Additionally, a region encoding functions related to cellobiose metabolism (LBA0871-LBA0883) was identified as absent from the commercial isolate sequence, L. acidophilus CIRM BIA-445 (Figure 3).
L. acidophilus clustered regularly interspaced short palindromic repeats (CRISPRs)
CRISPRs regions provide a unique insight into the evolution of bacterial phage resistance and have also been recently proposed as a means to identify industrial isolates24. One CRISPR region was identified within the L. acidophilus NCFM reference genome23 and at the genome scale this showed considerable synteny with all other L. acidophilus sequences investigated (Figure 3). To further interrogate small sequence changes in this region of the L. acidophilus genome assumed to be polymorphic as a result of historical phage attack, 20 genomes that contained a complete CRISPR region on a single sequence contig were compared (Supplementary Figure S2). The L. acidophilus NCFM CRISPR was defined as the archetypal reference sequence for this analysis and was composed 32 units of a repeat region and a spacer region23. The shortest CRISPR sequences were present in 5 culture collection isolates (L. acidophilus CIP 76.13T, LMG 13550T, ERR203994T, DSM 20242 and LAB 283), each of which were missing 3 spacer sequences (CRISPR types 4 and 6, Figure S2). CRISPR sequences from isolates re-sequenced under different aliases (DSM 20242 and LAB 283, CRISPR type 6 and 3 representing the type strain, CRISPR type 4; Figure S2) were conserved within-isolate, with the exception of L. acidophilus LMG 9433T, which possessed spacer sequences 2 and 3 (these were not present in CIP 76.13T, LMG 13550T or ERR203994T sequences; Figure S2). Nine of the 11 commercial isolates examined had identical CRISPR regions with no evidence of absent or duplicated spacers in relation NCFM (CRISPR type 3; Figure S2). The two commercial isolates with variant CRISPRs both possessed differences in relation to spacer 7. This spacer was duplicated after its first occurrence and absent at its third occurrence for isolate CulT2, while L. acidophilus C46 just lacked the third occurrence of spacer 7 (Figure S2). CRISPR sequence types (Figure S2) did not fit parsimoniously onto the whole genome phylogenetic network (Figure 2).
Conservation of L. acidophilus phenotypic traits
The lack of diversity seen within the L. acidophilus genome sequences suggested that the phenotypes of the corresponding isolates would also be invariant. Biochemical assessment (API 50CHL) of the carbohydrate fermentation profile of the L. acidophilus isolates (Table 1) was diagnostic of the species, but did not show significant differences between the commercial or culture collection isolates. The growth kinetics of selected commercial and culture collection isolates (Table 1) was also examined and no significant differences in lag phase, maximal growth rate and maximum culture density was seen (Figure S3). Finally, to examine the isolate phenotype at the protein level, Matrix-Assisted Laser Desorption/Ionization-Time-of-Flight Mass Spectrometry (MALDI-TOF MS) was carried out as a high-resolution analysis (Figure 4). While separation of L. acidophilus protein profiles generated by nine isolates (Table 1) from other LAB species –as shown in other studies25– was observed (Figure 4, Panel A), there was no differentiation by MALDI-TOF of the commercial and culture collection isolates (Figure 4, Panel B).

Diversity of L. acidophilus MALDI-TOF profiles.
MALDI-TOF profile distance scores were plotted in two dimensions as described in the Methods. Panel A shows the profiles of L. acidophilus (isolates NCFM, LMG 9433T, LMG 11428, LMG 11470, LMG 13550T, Rm 344, Rm 345, CUL 21 and CUL60; Table 1) compared to 6 other lactobacillus control species. Panel B shows the profiles of commercial and culture collection L. acidophilus Distances were calculated using the Pearson correlation similarity coefficient and position tolerance optimisation was set to 2%. Coordinates were calculated using multidimensional scaling.
Discussion
Numerous studies have used comparative genomics to identify similarities and differences within the LAB26,27,28 and for comparing species level diversity within the L. acidophilus group20, but to date no study has conducted a comparative genomics analysis encompassing a large number of LAB isolates below the species level. This represents a fundamental gap in knowledge concerning probiotic bacteria, as their beneficial characteristics may be unique to a single strain and encoded within their WGS. Understanding the genomics of LAB is also important from the regulatory perspective to accurately identify isolates and from the commercial standpoint to differentiate specific probiotic or fermentation traits. We have used WGS combined with a functional gene-by-gene diversity analysis approach to assess the infraspecies diversity of L. acidophilus as a single probiotic species. L. acidophilus was found to be a monophyletic species and isolates in global commercial use were clonal. The industrial significance of isolates with probiotic characteristics may have driven re-isolation and re-naming of the same isolate from environmental samples and little information is available concerning the history of proprietary commercial strains. Similar levels of genomic conservation were observed in the probiotic subspecies Bifidobacterium animalis subsp. lactis, where isolates from disparate commercial products were found to have highly conserved genome sequences and assumed to be an entirely monomorphic taxon, until the genome sequence of a culture collection isolate was found to represent a genomically unique strain29. These findings raise a number of questions relating to industrial strain identification, the commercial success of a single strain and whether human domestication has directed the evolution of L. acidophilus towards a narrow bottleneck.
Accurate identification of microbial content has been proposed as one of the most important product labelling criteria to support probiotic health claims30. The level of genetic conservation seen between probiotic L. acidophilus in this study and in previous analyses of two commercially distinct L. casei probiotic isolates31, could mean that probiotic health claims formulated from functional studies of one isolate could be applied to other, genetically monomorphic probiotic isolates. Multiple genetic and phenotypic identification strategies have been proposed as suitable to identify probiotic species, but no recommendations on how to analyse and interpret whole genome sequencing data have yet been proposed32. Although the 16S rRNA gene sequence has been widely used to classify Lactobacillus species19, given its conserved nature compared to other functional genes, traditional phylogenies drawn from the 16S rRNA gene sequence alone are often unstable and require addition of functional genes to improve resolution21,33. Our use of rMLST22 and wgMLST, implemented within BIGSdb18, was able to place L. acidophilus in the context of other L. acidophilus group members (Figure 1) and resolve strain differences within this essentially clonal species (Figure 2). Since multiple probiotic products are composed of mixtures microorganisms30, the utility of rMLST to resolve phylogenetic differences across domains22 makes it an ideal approach to bring unity and standardisation of strain identification to the probiotic field30.
Isolates investigated in this study showed no evidence of extrachromosomal DNA such as plasmids. Indeed, there was no evidence of assembling DNA beyond that homologous to the L. acidophilus NCFM reference sequence. Previously published L. acidophilus genomes15,16,23 also reflected this lack of plasmid DNA, although a single isolate – L.acidophilus 30SC – has two reported plasmids34. The anomalous presence of extrachromosomal DNA in the L. acidophilus 30SC genome sequence is due to the mis-identification of this strain; phylogenetic analysis of 30SC genome clearly demonstrates that it should have been classified as Lactobacillus amylovorus33,35. For this reason, the L. acidophilus 30SC genome sequence was excluded from analysis in this study. The correct identification of probiotic isolates, not only to satisfy product health claims and labelling guidelines, but also to maintain a rigorous standard for taxonomic assignment of genome sequences, is of particular importance for accurate downstream analysis.
Genome sequence analysis revealed that L. acidophilus as a bacterial species has remarkable genetic stability, especially when compared to other closely related Lactobacillus species (Figure 1). Going beyond this, the lack of variation among L. acidophilus isolates in commercial use is striking. One possible explanation for this is the global propagation, storage and repeated re-use of commercial probiotic isolates of L. acidophilus from within the commercial isolate cluster. In a similar case in a different probiotic species, two commercial isolates of L. casei, isolated directly from probiotic products produced by different companies were found to share a virtually identical genome sequence and encode a comparable exoproteome31. The L. casei data31 and our data suggest that human practice in terms of the use of probiotic LAB or dairy starter cultures may restrict the “natural” evolution of these bacteria, leading the widespread distribution and ultimately human consumption of highly clonal strains.
The commercial success of L. acidophilus may also be attributed to its genomic stability, allowing manufacturers to maintain good batch-to-batch quality control for probiotic manufacture and dairy fermentations. Phage spoilage of bacterial starter cultures is a major problem for the food industry24, but remarkable lack of variation within the L. acidophilus CRISPR may suggest it has not recently undergone substantial phage attack, although functional degradation of genes associated with the CRISPR region has been documented in L. acidophilus NCFM36. This may explain why CRISPR sequence types do not fit parsimoniously onto the wgMLST phylogenetic network. While the L. acidophilus genome contains phage-like remnants (eg. PAU regions)23, it does not encode an active prophage and while bacteriophage interactions with other L. acidophilus group species are widespread37, there are no recent report of phages active on validated L. acidophilus strains. The effective phage-resistance of L. acidophilus may also be a reason for its commercial success and widespread usage as stable and reliable commercial LAB species.
Bacterial pathogens such as Yersinia pestis38 and Mycobacterium tuberculosis39 show low genetic diversity concomitant with reaching an evolutionary bottleneck once within the human host. Our data suggest that L. acidophilus reached a similar evolutionary constriction that was associated with its historical human use in dairy fermentation and that is now being propagated by commercial and widespread probiotic use. Despite considerable effort to isolate L. acidophilus from non-human associated sources (MB, PhD Thesis Cardiff University), isolates from outside the dairy and probiotic industry were not identified, except from animals such as rats (Table 1), which are implicitly linked with human waste and activity. The lack of diversity within the L. acidophilus genome also suggests that genotyping methods that only sample a portion of the genetic content such as 16S rRNA gene sequencing19, PCR-fingerprinting13, or multilocus sequencing typing14 will not have sufficient resolution to support a specific health claim and its association to a given probiotic strain30. From a clinical perspective, the availability of WGS data has already advanced our understanding of pathogen population biology17. Continued development of analysis techniques, widespread availability of cost-effective sequencing and dissemination of bioinformatics expertise will assist in translating what we have learned from pathogenic systems into probiotic systems, potentially transforming the ways in which we regulate the manufacture and commercial use of microorganisms.
Methods
L. acidophilus isolates and genome sequences
L. acidophilus isolates and genomes were drawn from the following sources: 12 isolates of L. acidophilus were obtained from the culture collections of Belgian Coordinated Collection of Microorganisms (BCCM/LMG) and the University of Gent Laboratory for Microbiology; 5 genome sequences representative of culture collection strains were also obtained from the databases (Table 1; 17 genomes representative of culture collection isolates); 12 commercial isolates were obtained from probiotic products13, a probiotic supplier (Cultech Ltd., Port Talbot, UK) and T. Klaenhammer (North Carolina State University, Raleigh, NC, USA; a duplicate isolate of NCFM); the genomes of 4 additional commercial isolates were obtained from the databases (Table 1). Sequencing reads from one L. acidophilus genome sequence of unknown provenance were obtained from the NCBI short read archive and assembled into a draft genome sequence (Table 1) as described. The date of isolate recovery, its source and strain aliases were investigated and crosschecked where possible using the Strain Information Database (www.straininfo.net). Culture, storage and identification of L. acidophilus isolates was carried out as previously described23. Additional genomes from L. acidophilus group species were drawn from the DNA databases (Table S1).
Whole genome sequencing
Genomic DNA was extracted from the growth of single-colony inoculated L. acidophilus cultures with a Wizard genomic DNA purification kit (Promega, Southampton, United Kingdom). Genome re-sequencing was performed by the Oxford Genomics Centre, Wellcome Trust Centre for Human Genetics; www.well.ox.ac.uk/ogc). Briefly, Illumina multiplex libraries were generated from genomic DNA acoustically sheared to 200 to 300 bp using a Covaris E210 device. DNA fragments were end repaired and a 3′ nontemplate adenosine residue was ligated to the Illumina multiplexing adaptor oligonucleotide for sequencing. Libraries were pooled and analyzed together, in equiolar amounts, in a flow cell lane of the Illumina HiSeq 2000, generating 100-bp paired-end reads, which were deposited in the NCBI sequence read archive (SRA) with run accessions ERR386024 – ERR386044 and ERR386051 – ERR386052. Genome sequence data were assembled using Velvet version 1.2.10 shuffle and optimization scripts to create contigs with optimal parameters, with k-mer lengths between 83 and 95 bp40. Assembled data were deposited in the rMLST genome database (http://rmlst.org/), implemented with Bacterial Isolate Genome Sequence Database (BIGSdb) software18. The BIGSdb autotagger automatically identified rMLST loci, assigned alleles and tagged the sequences for future reference. The database automatically provided a report of the rMLST allelic profiles18.
rMLST and whole genome analysis
Relationships among L. acidophilus group isolates were established using phylogenetic networks based on rMLST sequences22. The 53 L. acidophilus group rps loci identified in the automated annotation process were compared among all isolates using the BIGSdb Genome Comparator module18. The aligned sequences were visualized with the Neighbor-net algorithm implemented in SplitsTree version 4.13.141. The vector graphics editor Inkscape 0.48.4 (www.inkscape.org) was used to annotate Neighbor Net images. L. acidophilus isolates were further analysed using whole genome MLST (wgMLST)18 at 1,864 loci defined in the genome sequence of L. acidophilus NCFM23 with the Genome Comparator. A distance matrix based on shared alleles was generated and visualised with NeighborNet18. Whole genome alignments were visualised and annotated using BLAST Ring Image Generator (BRIG) v0.9537.
CRISPR identification and analysis
L. acidophilus NCFM CRISPR sequence was used to search other L. acidophilus genome sequence data using BLAST+ tools implemented via the BIGSdb Web-interface18. The CRISPRtionary: Dictionary Creator tool at the CRISPRdb (http://crispr.u-psud.fr/crispr/)42 was used to identify direct repeat and spacer sequence in genomic regions containing CRISPR sequences, assigning a numerical value to each new spacer sequence encountered. The numerical profiles generated by CRISPR spacer sequences were compared. Each unique CRISPR sequence was assigned a CRISPR sequence type. Incomplete or partially assembled CRISPR regions were excluded from further analysis.
Phenotypic analysis
The phenotype of the L. acidophilus isolates (Table 1) and control species (L. brevis LMG 6906T, L. casei 6904T, L. gasseri LMG 9203T, L. johnsonii LMG 9436T, L. paracasei subsp. paracasei LMG 7955, L. plantarum LMG 6907T and Enterococcus faecium LMG 14205) was examined by API50 CHL biochemical analysis following the manufacturer's instructions and using their profile database (BioMerieux Marcy l'Etoile, France). A Bioscreen Microbiolgical Growth Analyser C (Labsystems, Finland) was used to determine the growth kinetics of selected L. acidophilus strains (Table 1) and a L. casei LMG 6904T control as described43 and the specific growth parameters calculated using the R statistical software module grofit44. MALDI-TOF analysis of the cellular proteins was carried out as described25,45, using a 4800 Plus MALDI TOF/TOF™ Analyzer (Applied Biosystems, Framingham, MA, USA). Quadruplicate cell extracts were evaluated for each isolate (Table 1) and the profile data exported to BioNumerics 6.0 (Applied-Maths, Sint-Martens-Latem, Belgium) to enable normalisation and cluster analysis as described45. Multi-dimensional scaling (MDS) was used to visualize the matrix of data similarities generated by BioNumerics 6.0.
References
Kandler, O. Carbohydrate metabolism in lactic acid bacteria. Antonie van Leeuwenhoek 49, 209–224, 10.1007/BF00399499 (1983).
de Vos, W. Systems solutions by lactic acid bacteria: from paradigms to practice. Microbial Cell Factories 10, S2 (2011).
Douillard, F. P. et al. Comparative genomic and functional analysis of Lactobacillus casei and Lactobacillus rhamnosus strains marketed as probiotics. Appl Environ Microbiol 79, 1923–1933 (2013).
Felis, G. E. & Dellaglio, F. Taxonomy of Lactobacilli and Bifidobacteria. Curr Issues Intestinal Microbiol 8, 44–61 (2007).
Euzeby, J. P. List of Bacterial Names with Standing in Nomenclature: a folder available on the Internet. Int J Syst Bacteriol 47, 590–592 (1997).
Mattarelli, P. et al. Recommended minimal standards for description of new taxa of the genera Bifidobacterium, Lactobacillus and related genera. Int J Syst Evol Microbiol 64, 1434–1451, d (2014).
Kleerebezem, M. & Hugenholtz, J. Metabolic pathway engineering in lactic acid bacteria. Curr Opinion Biotechnol 14, 232–237 (2003).
Sanders, M. E. Probiotics: Considerations for human health. Nutrition Reviews 61, 91–99 (2003).
Shah, N. P. Functional cultures and health benefits. Int Dairy J 17, 1262–1277 (2007).
Bull, M., Plummer, S., Marchesi, J. & Mahenthiralingam, E. The life history of Lactobacillus acidophilus as a probiotic: a tale of revisionary taxonomy, misidentification and commercial success. FEMS Microbiol Letts 349, 77–87 (2013).
Paineau, D. et al. Effects of seven potential probiotic strains on specific immune responses in healthy adults: a double-blind, randomized, controlled trial. FEMS Immunol Med Microbiol 53, 107–113 (2008).
Turroni, S. et al. Oxalate consumption by lactobacilli: evaluation of oxalyl-CoA decarboxylase and formyl-CoA transferase activity in Lactobacillus acidophilus. J Appl Microbiol 103, 1600–1609 (2007).
Mahenthiralingam, E., Marchbank, A., Drevinek, P., Garaiova, I. & Plummer, S. Use of colony-based bacterial strain typing for tracking the fate of Lactobacillus strains during human consumption. BMC Microbiol 9, 251 (2009).
Ramachandran, P., Lacher, D. W., Pfeiler, E. A. & Elkins, C. A. Development of a tiered multilocus sequence typing scheme for members of the Lactobacillus acidophilus complex. Appl Environ Microbiol 79, 7220–7228 (2013).
Stahl, B. & Barrangou, R. Complete Genome Sequence of Probiotic Strain Lactobacillus acidophilus La-14. Genome Announcements 1, 10.1128/genomeA.00376-13 (2013).
Falentin, H. et al. Draft Genome Sequences of Five Strains of Lactobacillus acidophilus, Strain CIP 76.13T, Isolated from Humans, Strains CIRM-BIA 442 and CIRM-BIA 445, Isolated from Dairy Products and Strains DSM 20242 and DSM 9126 of Unknown Origin. Genome Announcements 1, 10.1128/genomeA.00658-13 (2013).
Maiden, M. C. et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nat Rev Microbiol 11, 728–736 (2013).
Jolley, K. A. & Maiden, M. C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC bioinformatics 11, 595 (2010).
Salvetti, E., Torriani, S. & Felis, G. E. The Genus Lactobacillus: A Taxonomic Update. Probiotics Antimicrob Prot 4, 217–226 (2012).
Berger, B. et al. Similarity and differences in the Lactobacillus acidophilus group identified by polyphasic analysis and comparative genomics. J Bacteriol 189, 1311–1321 (2007).
Claesson, M. J., van Sinderen, D. & O'Toole, P. W. Lactobacillus phylogenomics - towards a reclassification of the genus. Int J Syst Evol Microbiol 58, 2945–2954 (2008).
Jolley, K. A. et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158, 1005–1015 (2012).
Altermann, E. et al. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. P Natl Acad Sci USA 102, 3906–3912 (2005).
Barrangou, R. & Horvath, P. CRISPR: new horizons in phage resistance and strain identification. Ann Rev Food Sci Technol 3, 143–162 (2012).
Doan, N. T. et al. Validation of MALDI-TOF MS for rapid classification and identification of lactic acid bacteria, with a focus on isolates from traditional fermented foods in Northern Vietnam. Letts Appl Microbiol 55, 265–273 (2012).
Coenye, T. & Vandamme, P. Extracting phylogenetic information from whole-genome sequencing projects: the lactic acid bacteria as a test case. Microbiology 149, 3507–3517 (2003).
Makarova, K. et al. Comparative genomics of the lactic acid bacteria. P Natl Acad Sci USA 103, 15611–15616 (2006).
O'Sullivan, O. et al. Comparative genomics of lactic acid bacteria reveals a niche-specific gene set. BMC Microbiol 9, Artn 50 Doi 10.1186/1471-2180-9-50 (2009).
Loquasto, J. R. et al. Bifidobacterium animalis subsp. lactis ATCC 27673 is a genomically unique strain within its conserved subspecies. Appl Environ Microbiol 79, 6903–6910 (2013).
Farnworth, E. R. The evidence to support health claims for probiotics. J Nutr 138, 1250s–1254s (2008).
Douillard, F. P. et al. Comparative genome analysis of Lactobacillus casei strains isolated from Actimel and Yakult products reveals marked similarities and points to a common origin. Microbial Biotechnol 6, 576–587 (2013).
Herbel, S. R., Vahjen, W., Wieler, L. H. & Guenther, S. Timely approaches to identify probiotic species of the genus Lactobacillus. Gut Pathog 5, Artn 27 Doi 10.1186/1757-4749-5-27 (2013).
Bull, M. J., Marchesi, J. R., Vandamme, P., Plummer, S. & Mahenthiralingam, E. Minimum taxonomic criteria for bacterial genome sequence depositions and announcements. J Microbiol Meth 89, 18–21 (2012).
Oh, S. et al. Complete genome sequencing of Lactobacillus acidophilus 30SC, isolated from swine intestine. J Bacteriol 193, 2882–2883 (2011).
Salvetti, E., Fondi, M., Fani, R., Torriani, S. & Felis, G. E. Evolution of lactic acid bacteria in the order Lactobacillales as depicted by analysis of glycolysis and pentose phosphate pathways. Systematic and applied microbiology 36, 291–305 (2013).
Stern, A., Keren, L., Wurtzel, O., Amitai, G. & Sorek, R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends in genetics : TIG 26, 335–340, 10.1016/j.tig.2010.05.008 (2010).
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L. & Beatson, S. A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, Artn 402 Doi 10.1186/1471-2164-12-402 (2011).
Achtman, M. Population structure of pathogenic bacteria revisited. Int J Med Microbiol 294, 67–73 (2004).
Namouchi, A., Didelot, X., Schock, U., Gicquel, B. & Rocha, E. P. After the bottleneck: Genome-wide diversification of the Mycobacterium tuberculosis complex by mutation, recombination and natural selection. Genome Research 22, 721–734 (2012).
Zerbino, D. R. Using the Velvet de novo assembler for short-read sequencing technologies. Current protocols in bioinformatics/editoral board, Andreas D. Baxevanis ... [et al.] Chapter 11, Unit 11 15, 10.1002/0471250953.bi1105s31 (2010).
Bryant, D. & Moulton, V. Neighbor-Net: An agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21, 255–265 (2004).
Grissa, I., Vergnaud, G. & Pourcel, C. CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucl Acids Res 36, W145–148, 10.1093/nar/gkn228 (2008).
Rushton, L. et al. Key role for efflux in the preservative susceptibility and adaptive resistance of Burkholderia cepacia complex bacteria. Antimicrob Agents Chemother 57, 2972–2980 (2013).
Kahm, M., Hasenbrink, G., Lichtenberg-Frate, H., Ludwig, J. & Kschischo, M. grofit: Fitting Biological Growth Curves with R. J Statistical Soft. 33, 1–21 (2010).
De Bruyne, K. et al. Bacterial species identification from MALDI-TOF mass spectra through data analysis and machine learning. Syst Appl Microbiol 34, 20–29 (2011).
Acknowledgements
This work was funded by a Biotechnology and Biological Sciences Research Council PhD studentship award to M.B. (Doctoral Training Grant BB/F016557/1), with additional CASE sponsorship from Cultech Ltd., Baglan, Wales, UK. We thank Todd R. Klaenhammer (North Caroline State University, Raleigh, NC, USA) for providing a reference isolate of L. acidophilus NCFM.
Author information
Authors and Affiliations
Contributions
All authors contributed to the work presented in this paper as follows. M.B. performed the majority of the research and data analysis. K.A.J., J.B. and M.C.J.M. assisted with genome sequencing and BIGSdb analysis and M.A. and P.V. contributed to the MALDI-TOF phenotype assessments. M.B., P.V., J.R.M. and E.M. contributed to the study design and strain resources. M.B. and E.M. wrote the first draft of the manuscript and all authors contributed to is revision, further analysis and written development.
Ethics declarations
Competing interests
M.B. 's industrial CASE PhD studentship was partly sponsored by the Cultech Ltd, who manufacture nutritional supplements including probiotic products; the remaining authors have no conflicts of interests to declare.
Electronic supplementary material
Supplementary Information
Supplementary Figures and Tables
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Bull, M., Jolley, K., Bray, J. et al. The domestication of the probiotic bacterium Lactobacillus acidophilus. Sci Rep 4, 7202 (2014). https://doi.org/10.1038/srep07202
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07202
This article is cited by
-
Strategies to display heterologous proteins on the cell surface of lactic acid bacteria using as anchor the C-terminal domain of Lactobacillus acidophilus SlpA
World Journal of Microbiology and Biotechnology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
