
Abstract
Small ruminants are important components in the livelihood of millions of households in many parts of the world. The spread of the highly contagious peste des petits ruminants (PPR) disease, which is caused by an RNA virus, PPRV, across Asia and Africa remains a major concern. The present study explored the evolutionary and epidemiological dynamics of PPRV through the analyses of partial N-gene and F-gene sequences of the virus. All the four previously described PPRV lineages (I-IV) diverged from their common ancestor during the late-19th to early-20th century. Among the four lineages, PPRV-IV showed pronounced genetic structuring across the region; however, haplotype sharing among the geographic regions, together with the presence of multiple genetic clusters within a country, indicates the possibility of frequent mobility of the diseased individuals across the region. The gradual decline in the effective number of infections suggests a limited genetic variation, which could be attributed to the effective vaccination that has been practiced since 1990s. However, the movement of infected animals across the region likely contributes to the spread of PPRV-IV. No evidence of positive selection was identified from this study.
Similar content being viewed by others


Introduction
Recognizing the importance of small ruminants (i.e., sheep and goat) in food security, socio-economic growth and the livelihood of millions of households predominantly in Africa and Asia as well as in many other parts of the world, the Food and Agriculture Organization (FAO) and World Organization for Animal Health (OIE) have prioritized for complete eradication of the devastating peste des petits ruminants (PPR) disease that causes a very high mortality among small ruminants across Asia and Africa1,2,3,4,5. This highly contagious viral disease is caused by a non-segmented negative-strand RNA virus, peste des petits ruminants virus (PPRV)1,4,5,6,7,8. The continuing spread of this devastating disease across Asia and Africa appears to be a serious threat to the emerging economy2,4,5. Hence, there is an increasing awareness among researchers and policy makers of the need to fight this livestock-killing disease at a regional and global level1,2,3,4,5,9,10.
PPR disease, also known as the goat plague, was first reported in 1942 in Côte d'Ivoire11 and has subsequently been reported with high prevalence from African and Asian countries as well as from the European part of Turkey1,4,5,9,12,13,14,15. Although small ruminants are the principal host of this paramyxovirus, PPRV has been reported to infect several other species, including cattle, buffalo, free-living bharals, camel, Asiatic lion and wild ungulates9,13,16, thus indicating a successful adaptation to a wide range of host species inhabiting different agro-eco-climatic zones. The PPRV genome is approximately 16 kb in length and encodes six structural proteins: the nucleocapsid (N), phosphoprotein (P), matrix (M), fusion (F), hemagglutinin (H) and polymerase (L) proteins, in the order of 3′-N-P-M-F-H-L-5′ and two non-structural proteins, C and V1,5. Based on the partial nucleotide sequence data of the N17 and F18 genes, PPRV isolates can be classified into four lineages: I, II, III and IV. Previous reports suggested an extent of geographic specificity of these four lineages. For instance, while lineages I and II, respectively are restricted to western and central Africa, lineage III is restricted to eastern Africa and the Arabian Peninsula1,5,9. Interestingly, lineage IV, which is also referred to as the Asian lineage, has a wide geographic coverage ranging from Southeast Asia to the Middle East and has more recently expanded into northern Africa5. Lineage IV has not only the predominant distribution across the region but also is gradually replacing lineage III in some parts of Africa5,19,20. Although the reason why lineage IV is more widespread than other lineages is unknown, one might speculate that lineage IV may have a selective advantage over other lineages. Under such circumstances, some amino acid residues in the PPRV-IV proteins are expected to evolve adaptively.
Despite the fact that live attenuated vaccines have been widely used to protect small ruminants against circulating PPRV1,3,7, the continuous spread of PPR disease indicates two possible hypotheses: (1) emergence of new PPRV strains with new genetic makeups and greater fitness in the face of vaccine-elicited protection (e.g., like other RNA viruses21,22); and (2) lapses in regulatory control that ultimately lead to movement of diseased/infected individuals across the region/state/country without proper monitoring and surveillance. Either of these two hypotheses can result in the spread of this highly contagious disease across a wide geographic range. If the first hypothesis holds with an emergence of new PPRV strains that is vaccine-driven or due to antigenic drift (e.g., Mumford et al.23), we expect to observe an increase in viral genetic diversity over time as well as footprints of positive selection in viral genes and proteins that play a crucial role in the successful adaptation to new environments, hosts and/or geographic regions. Thus, the central objective of this study is to test the two hypotheses through assessing whether past vaccination efforts have any effect on the viral genetic diversity and whether any of the protein-coding genes in the PPRV genome exhibit adaptive evolutionary changes.
Given the ongoing risk posed by the PPRV, particularly the rapid spread of the PPRV-IV lineage across Asia and Africa, it is crucial to determine whether past vaccination efforts have had any significant effect on the reduction of PPRV viral diversity over time. By performing a Bayesian coalescent analysis24,25,26 using serially sampled partial N- and F-gene sequence data and by performing site-specific maximum likelihood (ML) based selection analyses for each protein-coding gene of PPRV27,28, we report the evolutionary rate and date of emergence of the currently circulating PPRV, the patterns of selection pressures as well as gain important insights into the epidemiology of this highly contagious paramyxovirus.
Results
Dating the emergence of PPR viral lineages
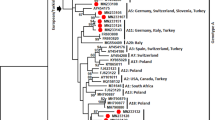
To infer the substitution rate and the time to the most recent common ancestors (TMRCAs) of PPRV lineages, we performed in-depth analyses using a Bayesian-based coalescent approach (Fig. 1A & B; Table 1). The Bayes factor (BF) statistic was used to select the best-fit clock model for the respective data sets. The BF is the difference in the marginal log likelihoods between two nested models: a model with the lowest marginal likelihood score (null model) and the other model with the highest marginal likelihood score (alternative model)29,30. For both N- and F- genes that comprised of all the four lineages and for the lineage IV of N-gene, the statistic of 2ln(BF) is greater than 20, thus providing strong evidence for a relaxed clock model with uncorrelated lognormal (UCLN) as the best-fit model (Table 1). The inferred substitution rates and TMRCAs for the respective data sets estimated under the best-fitting relaxed model that has the highest marginal log likelihood are shown in Table 1 and the inferred Maximum Clade Credibility (MCC) trees are shown in Figure 1. The MCC trees inferred from the partial N and F gene sequence data revealed the date of emergence of respective lineages of PPRV (Fig. 1A & B; Table 1). Both genes support the existence of four distinct lineages of PPRV (lineages, I-IV) with strong posterior probability support (≥ 0.99). Although both trees showed lineage IV and II clustered together, this inter-lineage clustering was weakly supported in the N-gene tree with posterior probability < 0.95 (Fig. 1A). Nevertheless, both genes showed that all the four lineages of PPRV diverged from a common ancestor. Subtle differences in the mean substitution rates of N- and F- genes of PPRV were observed (Table 1). The lower 95% Highest Posterior Density (HPD) value of Coefficient of Variation (CoV) was close to zero for the F gene, indicating no significant variations in evolutionary rate across the lineages (Table 1). In contrast, the higher CoV observed for the N gene and the 95% HPD not encompassing zero (Table 1) indicate large-scale variation in substitution rates across the lineages. The mean substitution rate for the N gene is higher than that of the F gene. Despite the subtle differences in substitution rates, both genes are consistent with a mean TMRCA of PPRV being around the late-1800s to the early-1900s (Table 1). Both genes also showed consistency in the time of divergence within respective lineages, which was estimated to have occurred between 1931 and 1990 (Fig. 1A & B). Separate analyses for lineage IV also showed that the substitution rate for the N gene is relatively higher than that of the F gene (Table 1). However, the TMRCA estimates from both genes indicate that the diversification within lineage IV was likely to have begun around early-to mid-1980s (Table 1).

Maximum clade credibility (MCC) trees from the Bayesian analysis of the PPRV partial (A) nucleocapsid and (B) fusion gene sequences.
The posterior probabilities and TMRCAs of the branches are depicted. Nucleotide accession number, country of origin and sampling year of each isolate is shown. Major nodes with weak posterior probability support (< 0.95) are indicated in asterisks (*).
Geographical spread of PPRV –IV
To assess whether each unique sequence (haplotype) of N-and F gene is specific to any geographic location or shared across the wide geographic range, we performed network analyses (Fig. 2A & B). From the 101 N-gene sequences and the 103 F-gene sequences, we obtained 56 (N1-N56) and 63 (F1-F63) haplotypes, respectively. In both genes, while most of the haplotypes showed geographic specificity, haplotypes N18, N10 (Fig. 2A) and haplotypes F46 and F34 (Fig. 2B) are shared among multiple countries. Although both genes showed an extent of geographic clustering of PPRV-IV (Fig. 2A & B), the clustering is more pronounced in the N-gene (Fig. 2A). In the N-gene network, PPRV-IV isolates from India, Bangladesh, Sudan and Turkey showed an existence of multiple clusters (Fig. 2A). Similarly, in the F-gene network, PPRV-IV isolates from Pakistan, India, Nepal, Sudan and Turkey also showed the existence of multiple clusters (Fig. 2B). The presence of multiple clusters within a country indicates either multiple waves of introduction of diseased animals from the neighboring countries or the recovery of viral populations from multiple bottlenecks. However, the closer association of a cluster with isolates from another country than with the clusters from the same country indicates that multiple waves of introduction of PPRV from different countries are more likely. For instance, in the N-gene network, the Indian isolates are more closely related with the isolates from Bangladesh in one cluster, whereas isolates from India and Israel are more closely related in another cluster (Fig. 2A). Similar patterns were also observed among the isolates from Pakistan, India, Turkey and Sudan in the F-gene network. Based on the results of the networks, it is likely that multiple haplotypes in individual countries and multiple countries connected by a single haplotype, supporting a relatively important trafficking of infected animals in Asia, Middle-East and more recently Africa. Due to the limited sample size from the respective countries, it was difficult to infer detailed migration and/or phylogeographic patterns. However, future studies, with larger sample size from respective countries, can be carried out for an in-depth understanding of the phylogeographic patterns of the PPRV-IV.

Parsimony networks based on the partial (A) N and (B) F genes of the PPRV isolates belong to lineage IV.
Each colored circle represents a unique gene sequence (haplotype) and the size of the circle is proportional to the number of isolates of that haplotype. Country of origin is indicated by unique color. Each empty circle between two adjacent colored circles represents the hypothetical mutational step not observed in the sample. All the isolates were connected at 95% confidence limit (connection limit = 7 mutations). The existence of multiple clusters in India and Pakistan are highlighted in N-and F genes, respectively.
Estimating the changing genetic diversity of PPRV-IV
The demographic history of the PPRV-IV was inferred using the Bayesian skyline plot (BSP) (Fig. 3). The effective number of infections estimated from the partial N and F genes showed consistent patterns (Fig. 3). The BSP showed three phases: a sudden increase in viral diversity during the year of 1987, followed by a stable phase until mid- to late-1990s, then followed by a gradual decline thereafter (Fig. 3). The sudden increase in viral diversity during 1987 could be associated with increases in host populations. The estimated genetic diversities for both genes are within the range of 10 to 100, which are consistent with the estimates reported for other paramyxoviruses31.

Bayesian skyline plots (BSP) depicting population dynamics of Asian lineage (lineage IV) of PPRV.
BSPs inferred from partial (A) N gene and (B) F gene depicting changing levels of genetic diversity (Neτ) of the PPRV-IV sampled between 1987 and 2011. The bold line represents the median estimate; the thin gray lines give the 95% HPD interval of the estimates.
Measuring selection pressure
Our analysis of selection pressure has revealed that the overall rate of nonsynonymous (dN) to synonymous (dS) substitution (ω = dN/dS) for each gene is less than 1 (Table 2), thus indicating purifying selection is the dominant force in the evolution of PPRV-IV. Although the overall ω indicated evidence of purifying selection, it is still difficult to infer whether any individual codons have evolved under positive selection. If a vast majority of codons have evolved under purifying selection and few codons were subjected to positive selection, the overall ω is expected to be less than 1 and the codons that have evolved under positive selection are likely to be undetected28. Considering this fact, we performed codon-specific substitution analyses that accounted for individual codon sites separately. Codon specific-analyses have revealed that, except the M protein, none of the genes showed evidence of positive selection (Table 2), thus indicating the mutations in the respective protein-coding genes are unlikely to have evolved adaptively. The biological significance of the single amino acid that was positively selected in the M protein is not known. Although the present analyses demonstrated the dominant role of purifying selection, sample size could possibly be a limiting factor and therefore, detection of positively selected sites from more samples representing different geographic regions and different hosts cannot be ruled out.
Discussion
The rapid spread of the highly contagious small ruminants-borne PPR viral disease across the Asian and African continents, which is costing the livelihood of millions of the poorest of the poor's living in the region, warrants urgent attention from the international community2,4. However, to design effective control measures, an in-depth understanding of the emergence of new viral strains and determining when, where and how these infectious viral pathogens cause the spread of infectious diseases is crucial32. Implementation of phylogenetic approaches to viral DNA sequence data is therefore imperative to elucidate the genetic relationships among different viral isolates sampled from different geographic and/or agro-eco climatic zones to provide important insights into their transmission and epidemiological dynamics. In the present study, we explored the evolutionary and epidemiological dynamics of PPRV through the analyses of the partial N-gene and F-gene sequences.
Previous epidemiological studies reported that PPR was first described in West Africa11 and subsequently, the disease was observed in other parts of Africa, followed by in the Middle East in 19831. Although these observations indicated the emergence of PPRV in 1940s, it is very difficult to predict the timing of the emergence of each lineage of PPRV from these previous epidemiological studies. Based on the Bayesian estimates, the currently circulating PPRV is estimated to have originated during the late 19th-to early 20th century and subsequently variants of lineages, I, II, III and IV began diverging during the early- to late-1900s. Consistently, previous studies reported that the TMRCA of the currently circulating measles viruses (MeV), a closely related virus to PPRV, was estimated to be within the last century (around 1889 to 1944)31,33. The divergence between MeV and RPV (rinderpest virus) was estimated have occurred around the 11th to 12th centuries; however, the effective population size at that time was sufficient for maintaining MeV33. Noting that estimated TMRCAs from sampled viral sequences can be very different from the time of origin indicated by historical data due to many reasons, notably, the effects of purifying selection31,34, the age of PPRV is probably older than what we estimated. Previous studies have demonstrated that branch lengths, whose accuracies are crucial in estimating the rates and TMRCAs, in viral phylogenetic trees, can potentially be underestimated due to purifying selection34. Therefore, estimation of intra and/or inter-species (for instance, PPRV, MeV and RPV) from the recently sampled viral pathogens may bias the TMRCA estimates34. Nevertheless, taking into account the codon-based substitution models, robust statistical approaches must be developed in order to have in-depth understanding of the ancient origin of the RNA viruses34.
Although the underlying genetic mechanisms that caused the evolution and emergence of these four lineages of PPRV are unclear, intuitively, the restricted distributions of lineages, I, II and III indicate that these lineages might have evolved through multiple founder events from a common ancestral population of PPRV in West Africa. The inadvertent introduction of the variants from one of these lineages in the Middle East, possibly through trade, might be a likely cause of the emergence of lineage IV through founder effect and subsequently spread across Asia and a recent re-introduction in Africa. Neutral theory of molecular evolution predicts that the effect of random genetic drift is likely to have a significant effect on small populations35, thus genetic accumulation could be extremely rapid in the small colonized viral populations that are founded by a few viral isolates32,36. Therefore, a founder effect could be a possible explanation for the emergence of these four lineages of PPRV at different time points.
The substitution rates inferred for PPRV are in the range of 10−3 to 10−4 subs/site/year and are compatible with the estimates of other RNA viruses, including the paramyxoviruses (e.g.,31,37,38). Such high substitution rates are typical characteristics of RNA viruses39; therefore, it does not necessarily mean high levels of antigenic variation31,37,39. The subtle differences in mutation rates among the genes could be associated with the function of the respective genes3 and level of selection pressures. The relatively higher mutation rate observed for nucleocapsid gene also reflected the pronounced genetic structuring of the PPRV-IV. Thus, in agreement with previous studies (e.g., Diallo et al.3), the present study suggested that N-gene could be a suitable marker for phylogenetic/population genetic studies of PPRV.
The Bayesian skyline plots revealed the population dynamics of PPRV-IV. Both N- and F-genes showed similar patterns, thus indicating both genes have evolved under similar selective pressures. The observed mutations accumulated in these genes are likely due to random genetic drift, which is evidenced by the codon-based selection analyses. Interestingly, the estimates of the viral genetic diversity are between 10 and 100, which are consistent with the estimates reported for other paramyxoviruses31. However, the estimates are significantly lower than those previously reported in chronic infections such as HIV40,41,42 and hepatitis C43,44. Such low levels of PPRV-IV diversity indicate that frequent population bottlenecks are likely to purge out the diseased individuals from the populations and therefore, limiting the genetic variation31. The gradual decline in PPRV-IV diversity since late-1990s could be associated with the effective preventive measures, including the mass vaccination programs adapted in different countries. Previous studies reported that the live attenuated vaccine derived from PPRV strain Nigeria 75/1, belonging to lineage II, was developed in 198945 and was successfully used to vaccinate more than 98,000 sheep and goats1,2. Subsequently, several vaccines, for instance, Arasur/87, Sungri/96 and Coimbatore/97 have been developed and successfully used in India and other countries worldwide (reviewed in8). Such effective vaccination campaign since 1990s could partly explain such a reduction in PPRV-IV viral diversity over time. Therefore, the spread of PPRV-IV infection due to the re-emergence of PPRV-IV resulting from the antigenic variation is highly unlikely. Although there is evidence that RNA viruses (e.g.,46,47) are prone to establish persistent infection through antigenic variation23,46, the lack of evidence of positive selection pressures in the viral proteins, notably in F and H genes that are crucial for successful viral adaptation in response to the host immune defense, together with the evidence of continual reduction in genetic diversity since the introduction of the mass vaccination program indicates that the observed patterns of diversity of PPR viruses are unlikely to be due to an antigenic drift.
Nevertheless, although the present study showed an overall reduction in PPRV-IV viral diversity, which indicates the efficacy of current vaccines used and/or correlating with generalized use of efficacious vaccines in some countries, the movement of diseased individuals, as revealed by genetic network analyses, could possibly contribute to the spread of the PPRV-IV virus across the Asian and African continents. Therefore, proper regulatory and bio-security measures should be adhered to in order to help control the spread of this disease.
Methods
Sequence data
A total of 131 N gene (255-bp) and 116 F gene (320-bp) partial nucleotide sequences representing diverse geographic regions and four lineages of PRRV were retrieved from GenBank48. The GenBank accession number and the year of isolation of each sequence are shown in Figure 1A & B. Sequences were aligned using MEGA version 449. Vaccine strains were excluded from the analyses.
Estimation of evolutionary parameters and population dynamics
We used a Bayesian Markov Chain Monte Carlo (MCMC) approach to estimate the overall substitution rate (measured in substitutions per site per year) for the N and F genes under the strict (constant molecular clock) and relaxed (uncorrelated lognormal, UCLN) molecular clocks under Bayesian skyline coalescent prior24,26 implemented in BEAST ver. 1.8.25. To select the best-fit evolutionary model for Bayesian MCMC inference, we performed a series of model selection analyses using BEAST. Different substitution and molecular clock models were compared by Bayes Factors (BF), which is the difference in the marginal log likelihoods between two nested models30. Large BF values, 2ln(BF) > 3 and >20, respectively indicate positive evidence and strong evidence against the null model (i.e., the model with the lowest marginal log likelihood)29. To estimate the evolutionary rates and infer the population dynamics, we used all the nucleotide sequences of the N (n = 131) and F (n = 116) genes whose years of isolation were available. The MCMC chains were run for sufficient time to achieve convergence. Phylogenies were evaluated using a chain length of 30 million states under a General Time Reversible (GTR) model with proportion of invariable sites (I) and gamma distribution shape parameter (G). Uncertainty in the data was described by the 95% highest posterior density (HPD) intervals. Multiple chains were run to access the convergence of trees and were checked using Tracer ver. 1.5 (available at: http://beast.bio.ed.ac.uk/Tracer). The inferred trees were visualized using FigTree ver. 1.4.1 (available at: http://tree.bio.ed.ac.uk/software/figtree/). We considered the node that has a posterior probability > 0.95 as with strong nodal support50. We utilized the Bayesian skyline plot (BSP)26 as a coalescent prior to infer the population history of PPRV-IV. The BSP estimates of effective population size of PPRV are measured in terms of relative genetic diversity. The changing genetic diversity can be quantified as Neτ, where Ne is the effective number of infections and τ is the infection-to-infection generation time31. Under relaxed molecular clock model (UCLN), large-scale variation of evolutionary rates in respective data sets were evaluated from the coefficient of variation (CoV) statistic, which represents the scaled variance in evolutionary rate among lineages24,29. The Maximum Clade Credibility (MCC) tree was generated using the TreeAnnotator software program implemented in the BEAST package.
Parsimony network
To know how the PPRV-IV isolates from different geographic regions are genetically connected, we performed parsimony network analyses using the viral DNA sequence data. We used partial nucleotide sequence data of the N and F genes to reconstruct the genealogical networks for the respective genes. The network was constructed at a 95% connection limit using TCS 1.2151.
Test for positive selection
Utilizing site-specific codon substitution models28, we determined whether any of the amino acid residues in the six protein-coding genes (i.e., N, P, M, F, H and L) of PPRV-IV are under positive selection. The complete nucleotide sequence data of the respective protein-coding genes were retrieved from GenBank and were used for selection analyses. Prior to selection analyses, a recombination detection program (RDP) implemented in the RDP ver 4.22 software package52 was used to determine if any of the genes showed evidence of recombination. The analyses revealed no evidence of recombination, thus allowing us to test for positive selection. We performed ML-based selection analysis to determine whether any of the genes are evolving under positive selection. Several codon-specific models (M7, M8 and M8a) implemented in the codeml program of PAML27 were used to determine which residues are evolving under positive selection. Site models allow the rate of nonsynonymous (dN) to the synonymous (dS) ratio (dN/dS = ω) to vary among residues. The input trees for selection analyses were reconstructed using the PhyML program53. The likelihood ratio test (LRT) was used to compare the M7 and M8a models that assume no positive selection (ω < 1) with the M8 model that assumes positive selection (ω > 1). Sites with Bayes Empirical Bayes (BEB) posterior probability ≥ 0.95 were considered to evolve under strong positive selection. Positively selected sites were also detected using the Fast Unbiased Bayesian Approximation (FUBAR) method54 via the Datamonkey website55. A posterior probability greater than 0.95 was used as the threshold for strong evidence of selection in FUBAR.
References
Albina, E. et al. Peste des Petits Ruminants, the next eradicated animal disease? Vet. Microbiol. 165, 38–44 (2013).
Diallo, A. Control of peste des petits ruminants and poverty alleviation? J. Vet. Med. B, Infect. Dis. Vet. public health 53 Suppl 1, 11–13 (2006).
Diallo, A. et al. The threat of peste des petits ruminants: progress in vaccine development for disease control. Vaccine 25, 5591–5597 (2007).
FAO. Supporting livelihoods and building resilience through Peste des Petits Ruminants (PPR) and small ruminant diseases control. Animal Production and Health Position Paper. Rome (2013). http://www.fao.org/docrep/017/aq236e/aq236e.pdf (Date of access: 29/01/2014)
Libeau, G., Diallo, A. & Parida, S. Evolutionary genetics underlying the spread of peste des petits ruminants virus. Anim. Front. 4, 14–20 (2014).
Diallo, A. Control of peste des petits ruminants: classical and new generation vaccines. Dev. Biol(Basel) 114, 113–119 (2003).
Parida, S. et al. Rescue of a chimeric rinderpest virus with the nucleocapsid protein derived from peste-des-petits-ruminants virus: use as a marker vaccine. J. Gen. Virol. 88, 2019–2027 (2007).
Sen, A. et al. Vaccines against peste des petits ruminants virus. Expert Rev. of Vaccines 9, 785–796 (2010).
Banyard, A. C. et al. Global distribution of peste des petits ruminants virus and prospects for improved diagnosis and control. J. Gen. Virol. 91, 2885–2897 (2010).
Baron, M. D., Parida, S. & Oura, C. A. Peste des petits ruminants: a suitable candidate for eradication? Vet. Rec. 169, 16–21 (2011).
Gargadennec, L. & Lalanne, A. La peste des petits ruminants. Bulletin des Services Zootechniques, et des Epizooties de I’ Afrique Occid ntale Francaise 5, 15–21 (1942).
Bao, J. et al. Complete genome sequence of a Peste des petits ruminants virus recovered from wild bharal in Tibet, China. J. Virol. 86, 10885–10886 (2012).
Bao, J. et al. Detection and genetic characterization of peste des petits ruminants virus in free-living bharals (Pseudois nayaur) in Tibet, China. Res. Vet. Sci. 90, 238–240 (2011).
Dilli, H. K., Geidam, Y. A. & Egwu, G. O. Peste de Petits Ruminants in Nigeria: A Review. Nigerian Vet. J. 32, 112–119 (2011).
Wang, Z. et al. Peste des petits ruminants virus in Tibet, China. Emerg. Infect. Dis. 15, 299–301 (2009).
Anderson, J. & McKay, J. A. The detection of antibodies against peste des petits ruminants virus in cattle, sheep and goats and the possible implications to rinderpest control programmes. Epidemiol. Infect. 112, 225–231 (1994).
Couacy-Hymann, E. et al. Rapid and sensitive detection of peste des petits ruminants virus by a polymerase chain reaction assay. J. Virol. Methods 100, 17–25 (2002).
Forsyth, M. A. & Barrett, T. Evaluation of polymerase chain reaction for the detection and characterisation of rinderpest and peste des petits ruminants viruses for epidemiological studies. Virus Res. 39, 151–163 (1995).
Kwiatek, O. et al. Asian lineage of peste des petits ruminants virus, Africa. Emerg. Infect. Dis. 17, 1223–1231 (2011).
Maganga, G. D. et al. Molecular typing of PPRV strains detected during an outbreak in sheep and goats in south-eastern Gabon in 2011. Virology J. 10, 82 (2013).
Dormitzer, P. R. et al. Influenza vaccine immunology. Immunol. Rev 239, 167–177 (2011).
Tusche, C., Steinbruck, L. & McHardy, A. C. Detecting patches of protein sites of influenza A viruses under positive selection. Mol. Biol. Evol. 29, 2063–2071 (2012).
Mumford, J. A. Vaccines and viral antigenic diversity. Rev. Sci. Tech. 26, 69–90 (2007).
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005).
Yang, Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
Yang, Z., Nielsen, R., Goldman, N. & Pedersen, A. M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155, 431–449 (2000).
Gray, R. R. et al. The mode and tempo of hepatitis C virus evolution within and among hosts. BMC Evol. Biol. 11, 131 (2011).
Suchard, M. A., Weiss, R. E. & Sinsheimer, J. S. Bayesian selection of continuous-time Markov chain evolutionary models. Mol. Biol. Evol. 18, 1001–1013 (2001).
Pomeroy, L. W., Bjornstad, O. N. & Holmes, E. C. The evolutionary and epidemiological dynamics of the paramyxoviridae. J. Mol. Evol. 66, 98–106 (2008).
Padhi, A. & Verghese, B. Molecular evolutionary and epidemiological dynamics of a highly pathogenic fish rhabdovirus, the spring viremia of carp virus (SVCV). Vet. Microbiol. 156, 54–63 (2012).
Furuse, Y., Suzuki, A. & Oshitani, H. Origin of measles virus: divergence from rinderpest virus between the 11th and 12th centuries. Virology J. 7, 52 (2010).
Wertheim, J. O. & Kosakovsky Pond, S. L. Purifying selection can obscure the ancient age of viral lineages. Mol. Biol. Evol. 28, 3355–3365 (2011).
Kimura, M. The Neutral Theory of Molecular Evolution. (Cambridge University Press, 1983).
Mayr, E. Change of genetic environment and evolution. (Allen and Unwin, 1954).
Holmes, E. C. The evolution and emergence of RNA viruses. (Oxford University Press, 2009).
Jenkins, G. M., Rambaut, A., Pybus, O. G. & Holmes, E. C. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54, 156–165 (2002).
Domingo, E. & Holland, J. J. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178 (1997).
Perez-Losada, M. et al. Phylodynamics of HIV-1 from a phase III AIDS vaccine trial in Bangkok, Thailand. PloS ONE 6, e16902 (2011).
Perez-Losada, M. et al. Phylodynamics of HIV-1 from a phase-III AIDS vaccine trial in North America. Mol. Biol. Evol. 27, 417–425 (2010).
Robbins, K. E. et al. U.S. Human immunodeficiency virus type 1 epidemic: date of origin, population history and characterization of early strains. J. Virol. 77, 6359–6366 (2003).
Magiorkinis, G. et al. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med. 6, e1000198 (2009).
Pybus, O. G. et al. The epidemic behavior of the hepatitis C virus. Science 292, 2323–2325 (2001).
Diallo, A., Taylor, W. P., Lefèvre, P. C. & Provost, A. Atténuation d'une souche de virus de la peste des petits ruminants: Candidat pour un vaccin homologue vivant. Rev. Elev. Med. Vet. Pays Trop. 42, 311–319 (1989).
Farci, P. et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288, 339–344 (2000).
Wang, J. H. et al. Characterization of antigenic variants of hepatitis C virus in immune evasion. Virology J. 8, 377 (2011).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–42 (2013).
Tamura, K., Dudley, J., Nei, M. & Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599 (2007).
Alfaro, M. E. & Holder, M. T. The Posterior and the Prior in Bayesian Phylogenetics. Annu. Rev. Ecol. Evol. Syst. 37, 19–42 (2006).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Martin, D. P. et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463 (2010).
Guindon, S. & Gascuel, O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Murrell, B. et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205 (2013).
Pond, S. L. & Frost, S. D. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533 (2005).
Acknowledgements
We thank Diana Chang for critical reading of this manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: A.P., L.M. Performed the experiments: A.P. Analyzed the data: A.P. Contributed reagents/materials: L.M. Wrote the paper: A.P., L.M.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
PPRV Nucleocapsid and Fusion gene sequence information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Padhi, A., Ma, L. Genetic and epidemiological insights into the emergence of peste des petits ruminants virus (PPRV) across Asia and Africa. Sci Rep 4, 7040 (2014). https://doi.org/10.1038/srep07040
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07040
This article is cited by
-
Paradigm shift in the diagnosis of peste des petits ruminants: scoping review
Acta Veterinaria Scandinavica (2020)
-
First detection and genetic characterization of peste des petits ruminants virus from dorcas gazelles “Gazella dorcas” in the Sudan, 2016-2017
Archives of Virology (2019)
-
Insight into the global evolution of Rodentia associated Morbilli-related paramyxoviruses
Scientific Reports (2017)
-
Genome sequencing of an Indian peste des petits ruminants virus isolate, Izatnagar/94, and its implications for virus diversity, divergence and phylogeography
Archives of Virology (2017)
-
First serological and molecular evidence of PPRV occurrence in Ghardaïa district, center of Algeria
Tropical Animal Health and Production (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
