
Abstract
Disruption of polyubiquitin gene Ubb leads to early-onset reactive gliosis and adult-onset hypothalamic neurodegeneration in mice. However, it remains unknown why reduced levels of ubiquitin (Ub) due to loss of Ubb lead to these neural phenotypes. To determine whether or not the defects in neurons or their progenitors per se, but not in their cellular microenvironment, are the cause of the neural phenotypes observed in Ubb−/− mice, we investigated the properties of cultured cells isolated from Ubb−/− mouse embryonic brains. Although cells were cultured under conditions promoting neuronal growth, Ubb−/− cells underwent apoptosis during culture in vitro, with increased numbers of glial cells and decreased numbers of neurons. Intriguingly, at the beginning of the Ubb−/− cell culture, the number of neural stem cells (NSCs) significantly decreased due to their reduced proliferation and their premature differentiation into glial cells. Furthermore, upregulation of Notch target genes due to increased steady-state levels of Notch intracellular domain (NICD) led to the dramatic reduction of proneuronal gene expression in Ubb−/− cells, resulting in inhibition of neurogenesis and promotion of gliogenesis. Therefore, our study suggests an unprecedented role for cellular Ub pools in determining the fate and self-renewal of NSCs.
Similar content being viewed by others


Introduction
Post-translational modification of proteins via ubiquitin (Ub) is known to play diverse functions, including proteasomal degradation, regulation of signal transduction pathways and endocytosis1,2,3. Since the discovery that neuron-specific protein gene product 9.5 (PGP9.5) is a Ub C-terminal hydrolase L14, ubiquitylation has also been shown to play a pivotal role in embryonic neural development5 and in neuronal function and dysfunction6, both of which require precise control of signaling pathways. In fact, ubiquitylation has been shown to be an essential process in neurogenesis, neuritogenesis and synaptogenesis and several E3 ligases, along with their specific substrates, have been identified in these processes5. However, the effect of reduced ubiquitylation of these substrates during neural development has not been elucidated.
We previously demonstrated that reduced levels of Ub due to loss of polyubiquitin gene Ubb leads to two striking neural phenotypes: adult-onset hypothalamic neurodegeneration and early-onset reactive gliosis7,8. Usually, neurodegeneration accompanies reactive gliosis, whereas in Ubb−/− mice, reactive gliosis precedes neurodegeneration in the hypothalamus. However, it remains unknown why reduced levels of Ub lead to reactive gliosis and it is unclear whether the defects reside in neurons or their progenitors per se, but not in their cellular microenvironment. To identify the exact cause of Ubb−/− neural phenotypes, we cultured cells isolated from embryonic brains to determine the presence of neuronal or neural stem/progenitor cell autonomous defects upon Ubb disruption.
During brain development, neural stem cells (NSCs) or progenitors generate neuroblasts and glioblasts, which differentiate into neurons and glial cells, such as astrocytes and oligodendrocytes, respectively9,10,11. The timing of these cell generation processes is important in the construction of normal brain cytoarchitecture and it is well known that neurogenesis precedes gliogenesis12. Usually, neurogenesis occurs during mid-gestation and ends at birth, whereas gliogenesis occurs mostly after birth13. One of the key regulators of this switch during NSC differentiation is Notch signaling14,15. Notch signaling is downregulated to promote neurogenesis in the embryonic stage and conversely, Notch signaling is upregulated to promote gliogenesis and maturation of neurons in the postnatal stage. In addition to the timed control of NSC differentiation, maintenance or self-renewal of NSCs has also been shown to be important for proper neural development16.
Here, we demonstrate that NSC numbers were reduced in Ubb−/− cells while glial cell numbers increased due to reduced proliferation of NSCs and to the dysregulated timing of gliogenesis. We hypothesize that NSCs in Ubb−/− mouse brains acquired higher gliogenic potential than neurogenic potential, meaning that gliogenesis occurred prematurely. Therefore, our data show that cellular Ub levels are important determinants in controlling the fate and timing of NSC differentiation into neurons and glial cells and cellular Ub levels also affect self-renewal of NSCs.
Results
Reduced survival and increased apoptosis in Ubb−/− cells isolated from embryonic brains
Cells were isolated from embryonic brains on 14.5 dpc (days post coitum) and cultured in medium to promote neuronal growth while suppressing growth of glial cells, such as astrocytes. Cultured wild-type cells displayed properties typical of neurons but not glial cells, as they were positive for the neuronal markers βIII-tubulin (TUJ1) and neuronal nuclei (NeuN), while negative for the astrocyte marker glial fibrillary acidic protein (GFAP) (Fig. 1a and 1b). On the other hand, cells were positive for GFAP and negative for TUJ1 and NeuN when cultured in medium that promotes glial cell growth (Fig. 1a and 1b). Therefore, cells were cultured in neuronal growth medium throughout the study.

Reduced number of Ubb−/− cells during culture in vitro.
(a) Immunoblot detection of neuronal marker TUJ1 and glial cell marker GFAP in cells isolated from wild-type (+/+) embryonic brains on 14.5 dpc that were cultured under two different medium conditions. Growth of neurons was facilitated in Neurobasal® medium with B-27 supplement (NB), whereas growth of glial cells was preferred in DMEM/10% FBS (DE). β-Actin (β-Act) was used as a loading control and mouse embryonic fibroblasts (MEFs) were included as a negative control. (b) Immunoblot detection of neuronal marker NeuN at 48 and 46 kDa in cells isolated from wild-type (+/+), Ubb+/− (+/−) and Ubb−/− (−/−) embryonic brains on 14.5 dpc and cultured in vitro for 13 days (DIV13). Wild-type (+/+) cells were cultured in two different medium conditions (NB and DE). (c) Determination of total cell numbers in Ubb+/+ (n = 19), Ubb+/− (n = 25) and Ubb−/− (n = 17) embryonic brains on 14.5 dpc. (d) Cells isolated from Ubb+/+ (n = 4) and Ubb−/− (n = 3) embryonic brains were plated on 96-well plate at 5 × 104 cells/well and cultured in vitro and counted using a hematocytometer. Representative immunoblot results of cells from two different embryonic brains per genotype are shown (a, b) and data are expressed as the means ± SEM from the indicated number of samples (c, d). **P < 0.01; ***P < 0.001 vs.Ubb+/+ (c) or Ubb+/+ on each day (d).
Since Ubb−/− mice are born smaller than their wild-type littermates8, the brains of Ubb−/− embryos were also smaller than those of wild-type embryos on most occasions (unpublished observation). Accordingly, the total cell number obtained from Ubb−/− embryonic brains was about 70% compared to wild-type on 14.5 dpc (Fig. 1c). Although we plated the same number of cells from wild-type and Ubb−/− embryonic brains, the intensity of NeuN-immunoreactive bands in the lysates of Ubb−/− cells cultured in vitro for 13 days was about 65% compared to wild-type cells (Fig. 1b). Therefore, we monitored the number of cells during the culture period. The total number of Ubb−/− cells continuously decreased during culture, whereas the number of wild-type cells was maintained (Fig. 1d).
To determine whether or not the gradual reduction of Ubb−/− cells during culture can be attributed to increased apoptosis, we performed TUNEL assay using cells cultured in vitro for 3 days, just before we observe the reduction of Ubb−/− cells. Although TUNEL+ cells were hardly observed in wild-type cells, one third of Ubb−/− cells were TUNEL+ (Fig. 2a). Thus, it is evident that the reduction in Ubb−/− cells was due to increased apoptosis. To identify Ubb−/− cell types that undergo apoptosis, cells cultured in vitro for 3 days were stained for GFAP, NeuN, or NSC marker nestin (NES) in combination with the TUNEL assay (Fig. 2b). Of the apoptotic cells (~34%), about 12% were GFAP+, about 11% were NeuN+ and about 8% were NES+ (Fig. 2c). Non-apoptotic GFAP+, NeuN+ and NES+ cells were 22%, 21% and 15%, respectively. Therefore, due to deletion of Ubb, all cell types that we investigated underwent apoptosis to some extent. However, it is highly likely that apoptotic stress is promoted in Ubb−/− cells during culture in vitro, as there was no evidence of apoptotic cell death in Ubb−/− embryonic brains on 14.5 dpc (Fig. 2d). This result also supports our previous observation that the number of hypothalamic neurons is maintained in 1-month-old Ubb−/− mice, while it is reduced by about 30% in adult Ubb−/− mice8.

Increased apoptosis in Ubb−/− cells during culture in vitro.
(a) Cells isolated from wild-type (Ubb+/+) and Ubb−/− embryonic brains (n = 3 each) on 14.5 dpc were cultured in vitro for 3 days. To determine the percentages of cells undergoing apoptosis, the total number of TUNEL+ cells was divided by the total number of DAPI+ cells in three randomly selected fields for each sample. For each sample, more than 100 DAPI+ cells were counted. (b) Cells isolated from wild-type (Ubb+/+) and Ubb−/− embryonic brains on 14.5 dpc and cultured in vitro for 3 days (DIV3) were subjected to a double-labeling fluorescence TUNEL assay with the NSC marker NES, neuronal marker NeuN and glial cell marker GFAP and DNA was visualized with DAPI. Arrowheads indicate TUNEL+ apoptotic cells that are also positive for cell-type specific markers. (c) The percentage of apoptotic (TUNEL+) or non-apoptotic (TUNEL−) NES+, NeuN+, or GFAP+Ubb−/− cells (n = 3) was determined in a similar manner as described in (a). (d) Immunoblot detection of apoptotic marker caspase-3 (CASP3) in wild-type (+/+) and Ubb−/− (−/−) embryonic brain lysates on 14.5 dpc (14.5 dpc) or in cells isolated from embryonic brains on 14.5 dpc and cultured in vitro for 7 days (DIV7). CASP3-immunoreactive bands were detected at 35 and 17 kDa, representing pro-caspase-3 (pro-CASP3) and active caspase-3 (active CASP3). β-Actin (β-Act) was used as a loading control. (e) Wild-type (Ubb+/+) and Ubb−/− cells on DIV13 were stained with markers for GFAP or NeuN and DNA was visualized with DAPI. (f) On DIV3 and DIV13, the percentages of wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) positive for NeuN or GFAP were determined in a similar manner as described in (a). Representative images or immunoblot results of cells from three different embryonic brains per genotype are shown (b, d, e) and data are expressed as the means ± SEM from the indicated number of samples (a, c, e). *P < 0.05; **P < 0.01; ***P < 0.001 vs.Ubb+/+ on each day or as indicated by bars. Scale bars, 50 μm.
Intriguingly, when Ubb was disrupted, the number of GFAP+ astrocytes gradually increased during culture (Fig. 2e and 2f), suggesting that the number of proliferating GFAP+ cells exceeds that of apoptotic GFAP+ cells. It took for a week for newly attached GFAP+ cells to obtain their distinguishable morphology of astrocytes when cultured in neuronal growth medium (see Fig. 2b and 2e). On the other hand, NeuN+ neuronal populations in Ubb−/− cells remained significantly lower than those in wild-type cells throughout the culture period (Fig. 2e and 2f). Therefore, apoptosis in Ubb−/− cells may be closely associated with the increase in the number of glial cells.
Reduced neural stem cell numbers and dysregulation of their differentiation upon disruption of Ubb
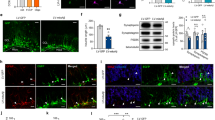
As the phenotype of Ubb−/− cells was apparent on DIV3, defects in Ubb−/− cells might already have been present at the beginning of culture. Of interest, expression levels of Gfap, but not Fox3, which was identified as NeuN17, significantly increased in Ubb−/− cells from DIV7 to DIV13. Therefore, we investigated the dysregulation of NSC differentiation into neurons and glial cells, which is thought to be tightly controlled during embryonic brain development12. To determine whether disruption of Ubb affects NSC differentiation, we measured the percentage of NES+ NSCs in a given cell population on DIV1 (Fig. 3a and 3b). To our surprise, the percentage of NES+ NSCs significantly decreased in Ubb−/− cells, even on DIV1, accompanied by a significant decrease in NeuN+ neurons, as well as by a significant increase in GFAP+ glial cells (astrocytes) (Fig. 3a and 3b). Decreased number of NES+ NSCs and increased number of GFAP+ astrocytes were also evident in Ubb−/− embryonic brains on 14.5 dpc (Fig. 3d).

Reduced number of neural stem cells and dysregulation of their differentiation in Ubb−/− cells.
(a) Cells isolated from wild-type (Ubb+/+) and Ubb−/− embryonic brains on 14.5 dpc and cultured in vitro for 1 day (DIV1) were stained with the NSC marker NES, neuronal marker NeuN and glial cell marker GFAP in combination with a proliferation marker PH3 and DNA was visualized with DAPI. Arrowheads indicate PH3+ cells that are also positive for cell-type specific markers. (b) On DIV1, the percentages of wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) positive for NES, NeuN, or GFAP were determined in a similar manner as described in Figure 2(a). (c) On DIV1, the percentages of PH3+ wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) that were also positive for NES, NeuN, or GFAP were determined in a similar manner as described in Figure 2(a). (d) Frozen brain sections were prepared from wild-type (Ubb+/+) and Ubb−/− embryos on 14.5 dpc and stained with the NSC marker NES and glial cell marker GFAP and DNA was visualized with DAPI. (e) Nes, Fox3 (NeuN) and Gfap mRNA levels in cells isolated from wild-type (Ubb+/+) and Ubb−/− embryonic brains (n = 3 each) on 14.5 dpc (DIV0) or cultured in vitro for 1 day (DIV1) were determined by qRT-PCR and normalized to Gapdh levels. mRNA levels are expressed as a fold increase relative to wild-type levels on DIV0. Representative images of cells or sections from three different embryonic brains per genotype are shown (a, d) and data are expressed as the means ± SEM from the indicated number of samples (b, c, e). *P < 0.05; **P < 0.01; ***P < 0.001 vs.Ubb+/+ on each day. Scale bar for (a), 50 μm; scale bar for (d), 500 μm.
To determine whether the decreased number of NES+ NSCs in Ubb−/− cells was due to the reduced proliferation of NSCs, cells were co-stained with a proliferation marker, phospho-histone 3 (PH3) (Fig. 3a). We observed a dramatic reduction in the proliferation of NSCs and a significant increase in the proliferation of astrocytes in Ubb−/− cells (Fig. 3a and 3c). These results suggest that NSC cell numbers are intrinsically low in Ubb−/− embryonic brains due to reduced capacity to self-renew or proliferate to maintain NSCs. In NeuN+ neurons, genes related to neuronal maturation have been shown to be transcriptionally activated by acetylation of Lys14 in H3, which facilitates the phosphorylation of H318,19. Therefore, the occasionally observed PH3 immunoreactivity in NeuN+ neurons may not necessarily indicate the proliferation status of cells.
To further confirm these results, we measured the expression levels of Nes, Fox3 (NeuN) and Gfap in cells isolated from embryonic brains on both 14.5 dpc (no culture in vitro; DIV0) and DIV1 using qRT-PCR (Fig. 3e). In accordance with the immunofluorescence results, expression levels of Nes and Fox3 (NeuN) significantly decreased, while Gfap expression significantly increased in cells isolated from Ubb−/− embryonic brains on 14.5 dpc, even before the start of in vitro culture. Similar results were obtained when we measured the mRNA levels of Nes, Fox3 (NeuN) and Gfap in embryonic brains on 14.5 dpc (Supplementary Fig. S1a). These results suggest that differentiation of NSCs into neurons during mid-gestation was somehow inhibited, whereas premature NSC differentiation into glial cells occurred upon disruption of Ubb. This latter phenomenon is known to occur mostly after birth13. Therefore, the decrease in the number of NSCs in Ubb−/− embryonic brains can also be attributed to the dysregulation of NSC differentiation.
Uprgulation of Notch target genes in Ubb−/− cells due to increased steady-state levels of NICD
The timing of NSC differentiation into neurons (neurogenesis) or glial cells (gliogenesis) is controlled by Notch signaling. Specifically, differentiation into neurons is promoted by repression of Notch signaling during the mid-gestation period of embryonic development15. On the other hand, activation of Notch signaling after birth promotes differentiation into glial cells. Notch activation also supports the maturation of already differentiated neurons15. Therefore, we measured the expression levels of Notch along with its target genes in cells isolated from wild-type and Ubb−/− embryonic brains on both 14.5 dpc (DIV0) and DIV1. Expression levels of Notch, as well as its target genes, significantly increased in Ubb−/− cells (Fig. 4a) and remained high in Ubb−/− cells throughout the culture period (Fig. 4b). Similar results were obtained when we measured the mRNA levels of Notch and its target genes in embryonic brains on 14.5 dpc (Supplementary Fig. S1b).

Upregulation of Notch target genes and increased steady-state levels of NICD in Ubb−/− cells.
(a) Notch1, Hes5 and Hey1 mRNA levels in cells isolated from wild-type (Ubb+/+) and Ubb−/− embryonic brains (n = 3 each) on 14.5 dpc (DIV0) or cultured in vitro for 1 day (DIV1) were determined by qRT-PCR and normalized to Gapdh levels. mRNA levels are expressed as a fold increase relative to wild-type levels on DIV0. (b) Expression levels of Notch and its target genes in wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) on DIV7 and DIV13 were determined by qRT-PCR and normalized to Gapdh levels. mRNA levels are expressed as a fold increase relative to wild-type levels on DIV7. To repress Notch signaling, Ubb−/− cells cultured in vitro for 5 days (DIV5) were treated with 10 μM DAPT and cultured for another 2 to 8 days (DIV7 and DIV13). One-half of the medium containing 10 μM DAPT was changed every other day. (c) Cycloheximide (CHX) chase of stabilized NICD. Cells isolated from wild-type (+/+) and Ubb−/− (−/−) embryonic brains (n = 3 each) on 14.5 dpc and cultured in vitro for 7 days (DIV7) were pre-treated with 10 μM MG-132 for 2 hr to prevent degradation of NICD. Upon removal of MG-132, accumulated NICD was chased in medium containing 10 μg/ml CHX for up to 6 hr. β-Actin (β-Act) was used as a loading control. (d) Immunoblot detection of cleaved NICD in cells isolated from wild-type (+/+) and Ubb−/− (−/−) embryonic brains on 14.5 dpc and cultured in vitro for 7 and 13 days (DIV7 and DIV13). Representative immunoblot results of cells from three different embryonic brains per genotype are shown (c, d) and data are expressed as the means ± SEM from the indicated number of samples (a–c). #P < 0.1; *P < 0.05; **P < 0.01; ***P < 0.001 vs.Ubb+/+ on each day or as indicated by bars.
To confirm that the upregulation of Notch target genes is indeed due to activation of Notch signaling in Ubb−/− cells, pharmacological intervention of Notch signaling using DAPT, a well-known inhibitor of γ-secretase, which indirectly inhibits Notch signaling by hampering cleavage of NICD, was exploited during culture in vitro. Upon treatment with DAPT, expression levels of Notch, as well as its target genes in Ubb−/− cells, decreased to those of wild-type cells (Fig. 4b). These results suggest that the upregulation of Notch target genes in Ubb−/− cells can be related to levels of cleaved NICD. To investigate this possibility, we measured the steady-state levels of Notch intracellular domain (NICD), which was released following endocytosis of the Notch ligand Delta as well as the extracellular region of the Notch receptor in neighboring cells20,21. Increased steady-state levels of cleaved NICD were observed in Ubb−/− cells, probably due to the prolonged stabilization of NICD, which is usually rapidly degraded by the proteasome (Fig. 4c and 4d). Therefore, aberrant activation of Notch signaling in Ubb−/− cells is due to increased steady-state levels of NICD, although we cannot exclude the possibility that increased expression of Notch itself may also contribute to increased levels of NICD.
Aberrant activation of Notch signaling in Ubb−/− cells is linked to premature onset of gliogenesis and suppression of neurogenesis
Notch target genes Hes5 and Hey1, which encode inhibitory basic helix-loop-helix (bHLH) transcription factors, are known to repress the function of proneuronal bHLH transcription factors encoded by proneuronal genes, such as Neurog122. Neurogenins are required for activation of neuronal genes as well as neuronal differentiation. As expected, Neurog1 expression levels were significantly reduced in Ubb−/− cells on DIV0 and DIV1, as well as in embryonic brains on 14.5 dpc (Fig. 5a and Supplementary Fig. S1c) and reduced Neurog1 expression was also observed in Ubb−/− cells throughout the culture period (Fig. 5b). Therefore, reduced expression of proneuronal genes in Ubb−/− embryonic brains can directly contribute to the inhibition of neuronal differentiation and the promotion of glial differentiation.

Improvement of Ubb−/− neuronal and glial phenotypes via pharmacological intervention of Notch signaling.
(a) Expression levels of the proneuronal gene Neurog1 in wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) on DIV0 and DIV1 were determined by qRT-PCR as described in Figure 4(a). (b) Expression levels of Neurog1, Fox3 (NeuN) and Gfap in wild-type (Ubb+/+) and Ubb−/− cells (n = 3 each) on DIV7 and DIV13 were determined by qRT-PCR as described in Figure 4(b). DAPT treatment of Ubb−/− cells was carried out as described in Figure 4(b). (c) The percentage of apoptotic (TUNEL+) or GFAP+ cells in wild-type (Ubb+/+), Ubb−/− and DAPT-treated Ubb−/− cells (n = 3 each) on DIV7 and DIV13 was determined in a similar manner as described in Figure 2(a). (d) Immunoblot detection of TUJ1 in wild-type (Ubb+/+), Ubb−/− and DAPT-treated Ubb−/− cells on DIV7 and DIV13. β-actin (β-Act) was used as a loading control. Representative immunoblot results of cells from three different embryonic brains per genotype are shown (d) and data are expressed as the means ± SEM from the indicated number of samples (a–c). #P < 0.1; *P < 0.05; **P < 0.01; ***P < 0.001 vs.Ubb+/+ on each day or as indicated by bars.
To confirm that aberrant activation of Notch signaling in Ubb−/− cells is responsible for inhibition of neurogenesis and promotion of gliogenesis, expression levels of Neurog1, Fox3 (NeuN) and Gfap were determined in the presence or absence of DAPT. As expected, Fox3 (NeuN) mRNA levels decreased while Gfap mRNA levels increased in Ubb−/− cells compared to wild-type cells during the culture period (Fig. 5b). However, upon repression of Notch signaling by DAPT, Neurog1 and Fox3 (NeuN) mRNA levels increased significantly, while Gfap mRNA levels were reduced significantly in Ubb−/− cells on DIV13 (Fig. 5b). In addition, a significant reduction of apoptosis was detected in Ubb−/− cells during culture in the presence of DAPT (Fig. 5c). Repression of Notch signaling also decreased the number of GFAP+ astrocytes and increased levels of TUJ1+ neurons during the culture period (Fig. 5c and 5d). Based on these results, we conclude that aberrant activation of Notch signaling in Ubb−/− cells is, at least in part, responsible for dysregulation of NSC differentiation with premature onset of gliogenesis and suppression of neurogenesis.
Discussion
In this study, we showed that cellular Ub pools determine the differentiation of NSCs into neurons or glial cells via regulation of Notch signaling (Fig. 6). In the embryonic stage, Notch signaling is downregulated, promoting neurogenesis and suppressing gliogenesis. In the postnatal stage, Notch signaling is upregulated, which supports gliogenesis and neuronal maturation. Since Notch activation is required for self-renewal or maintenance of NSCs, timely and sustained downregulation of Notch signaling is an essential determinant of neurogenesis during the mid-gestation period14. After Notch receptor interacts with its ligand Delta (DLL1) in neighboring cells, Notch signaling is initiated by cleavage of NICD by γ-secretase23. Released NICD then migrates into the nucleus and functions as a part of the transactivator complex to promote transcription of Notch target genes. Therefore, timely degradation of NICD is likely important for tight regulation of Notch signaling.

Proposed model of Ub-dependent neural development via regulation of Notch signaling.
Disruption of polyubiquitin gene Ubb reduces levels of cellular Ub pools, resulting in prolonged activation of Notch signaling and impaired neural stem cell (NSC) maintenance or self-renewal. Notch activation during the embryonic stage could lead to dysregulation of NSC differentiation with premature gliogenesis and impaired neurogenesis. Defective neurons may be responsible for activation of astrocytes, which probably cause degeneration (deg.) of defective neurons. Detailed descriptions are provided in the Discussion section.
Decreased availability of cellular Ub pools due to disruption of polyubiquitin gene Ubb has prevented the timely degradation of NICD, resulting in increased transcription of Notch target genes. Because increased steady-state levels of NICD should definitely contribute to upregulation of Notch target genes, it is suspected that timely downregulation of Notch signaling may not be achieved during mid-gestation when Ub is not readily available, leading to prolonged activation of Notch signaling, which is the major cause of the dysregulated NSC differentiation.
To the best of our knowledge, our study is the first to demonstrate that aberrant activation of Notch signaling due to reduced levels of Ub in cells from mid-gestation embryonic brains can lead to alterations in the fate of NSCs, resulting in the premature onset of gliogenesis and suppression of neurogenesis. Furthermore, our results suggest that aberrant activation of Notch signaling may continue until the postnatal stage in Ubb−/− mice. Under high gliogenic potential, astrocytes dramatically increase in number and become reactive after losing neuroprotective functions or after gaining neurotoxic characteristics23,24. In fact, pro-inflammatory cytokine levels gradually increased in Ubb−/− cells during the culture period (Park et al., unpublished data). Therefore, early-onset reactive gliosis in Ubb−/− mice is most likely due to dysregulation of NSC differentiation during brain development resulting from the continued activation of Notch signaling.
Although we cannot completely exclude the possibility that the increased Notch expression in Ubb−/− cells can also be attributed to the increased astrocyte numbers or to the increased ratio of astrocytes to neurons, the following evidence suggests that this may not be the case. Based on the transcriptome database for astrocytes and neurons isolated from postnatal mouse brains on day 7 (P7), Gfap expression is about >100-fold higher in astrocytes than in neurons and Notch expression is about 10-fold higher in astrocytes than in neurons25. Between DIV0 and DIV1, the fold increases in Gfap and Notch expression in Ubb−/− cells compared to wild-type cells were almost similar (see Fig. 3e and 4a). Therefore, increased Notch expression was probably not due to the increased number of astrocytes. Although astrocyte numbers and Gfap expression increased dramatically in Ubb−/− cells as culture progressed (see Fig. 2f, 5b and 5c), there was no proportional increase in Notch expression (see Fig. 4b). Therefore, Notch activation in Ubb−/− embryonic brains and Ubb−/− cells may not be attributed to the presence of astrocytes, but the activation more likely causes premature differentiation of NSCs into astrocytes.
Although Notch activation is required for neuronal maturation, as Ubb−/− neurons were not properly generated during the embryonic stage, defective Ubb−/− neurons may not mature as wild-type neurons (Fig. 6). Defective neurons interact with and activate astrocytes, which, in turn, are responsible for the degeneration of these defective neurons or their progenitors23. One possible mechanism for this process is the secretion of lipocalin 2 (LCN2) by reactive astrocytes24, which exerts specific toxicity on neurons to promote their degeneration. LCN2 has also been known as a mediator of reactive astrocytosis and its expression is increased when astrocytes are under inflammatory stress or during reactive astrocytosis26. LCN2 is also responsible for upregulation of Gfap, which occurs during reactive astrocytosis. As Lcn2 mRNA levels increased dramatically in Ubb−/− cells during the culture period (Park et al., unpublished data), it is highly likely that Ubb−/− astrocytes underwent activation. In fact, adult-onset hypothalamic neurodegeneration in Ubb−/− mice is preceded by neuronal dysfunction in the hypothalamus7,8. Therefore, these dysfunctional neurons may be responsible for the activation of astrocytes, even though their numbers are maintained and the reactive astrocytes may be responsible for adult-onset neurodegeneration in Ubb−/− mice.
It was expected that aberrant activation of Notch signaling in Ubb−/− embryonic brains would enhance proliferation of NSCs. However, our data suggest that the capacity to maintain NSCs may be reduced in Ubb−/− embryonic brains due to reduced proliferation of NSCs. In fact, reduced cellular proliferation capacity is observed in various cell types when cellular Ub pools are reduced, including hematopoietic (stem) cells, fetal liver epithelial progenitor cells, hepatocytes and mouse embryonic fibroblasts27,28,29. The proliferation capacity of NSCs was also compromised in Ubb−/− embryonic brains, resulting in reduction of NSC numbers.
Expression levels of Notch ligand Dll1, Notch target gene Hes1 and proneuronal gene Neurog2 in NSCs have been shown to oscillate (repeated up and downregulation with 2–3 hour intervals) to maintain the status of NSCs before differentiation into neurons16,30. In particular, both Hes1 mRNA and HES1 protein levels oscillate in response to negative autoregulation of Hes1 transcription and Ub-dependent proteasomal degradation of HES1 protein, respectively31. Therefore, timely degradation of HES1, as well as other Notch signaling molecules in oscillations, including NICD, may not be tightly regulated when cellular Ub levels are reduced, resulting in the reduced capacity to maintain NSCs in Ubb−/− embryonic brains. These observations further support the finding that self-renewal or maintenance of NSCs is significantly reduced in Ubb−/− embryonic brains.
In conclusion, our study using cells isolated from mouse embryonic brains suggests that cellular Ub pools play an important role in determining the fate and self-renewal of NSCs. Decreased availability of cellular Ub pools resulted in reduced numbers of NSCs and these NSCs underwent premature gliogenesis and suppression of neurogenesis due to prolonged activation of Notch signaling, leading to defective neuronal development. Defective neurons may activate astrocytes into reactive astrocytes, resulting in apoptosis in defective neurons or their progenitors and even resulting in apoptosis in astrocytes. Therefore, our in vitro cell culture results support the phenotypes observed in Ubb−/− mice, including reactive gliosis and neurodegeneration7,8.
Methods
Mouse studies
All mice were maintained in plastic cages with ad libitum access to food and water, with a 12-hr light cycle32. Wild-type and Ubb−/− embryos were obtained from interbreeding of Ubb+/−(eGFP-puro) mice. All experiments were performed in accordance with relevant guidelines and regulations approved by the University of Seoul Institutional Animal Care and Use Committee (UOS IACUC). All experimental protocols, including breeding, euthanasia and dissection of embryonic brains, were approved by the UOS IACUC (approval numbers are UOS-091201-1 and UOS-121025-2).
Primary cell culture
Embryonic brains were dissected from 14.5 days post coitum (dpc) and placed in Ca2+ and Mg2+-free Hank's Balanced Salt Solution (HBSS) to remove the cerebellum and meninges. Isolated brains were transferred to trypsinization solution containing 0.05% trypsin/EDTA (Cellgro) and incubated for 30 min at 37°C with vigorous shaking at 250 rpm. Trypsinization was quenched by adding an equal volume of cell culture medium (DMEM supplemented with 10% fetal bovine serum (FBS), 20 mM L-glutamine and 1% antibiotics/antimycotics (Cellgro)). After centrifugation at 1,000 rpm for 5 min, the tissue was triturated in neuronal cell culture medium (Neurobasal® medium supplemented with B-27® supplement (Invitrogen), 1× GlutaMax, 0.5 mM L-glutamine and 1% antibiotics/antimycotics (Cellgro)) by gentle pipetting through a 1000 μl pipette tip. Triturated tissues were strained through a 40 μm nylon mesh. Resulting cells were plated on a tissue culture dish coated with poly-D-lysine (MW 30,000–70,000, Sigma-Aldrich) and laminin (Invitrogen) at 1 × 104 to 1.5 × 105 cells/cm2 depending on the experiment and then cultured in the same medium. One-half of the medium was changed every three days.
Immunofluorescence analysis and TUNEL assay
For immunofluorescence, cells grown on poly-D-lysine-coated coverslips were fixed in 4% paraformaldehyde for 10 min at room temperature (RT), permeabilized with 0.3% Triton X-100/PBS and blocked with 3% BSA/PBS for 1 hr at RT. Fixed cells were incubated with anti-GFAP (1:1,000, Millipore), anti-NeuN (1:200, Millipore), anti-NES (1:1,000, Abcam) or anti-PH3 (1:200, Millipore) antibody at 4°C overnight, washed with PBS and incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG or Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:1,000, Invitrogen) along with 0.1 μg/ml of DAPI for 1 hr at RT. Cells were then mounted using Prolong Gold antifade reagent (Invitrogen). Images were visualized with a Carl Zeiss AxioImager A2 microscope or Carl Zeiss Axiovert 200 M microscope equipped with a confocal laser scanning module LSM510. Immunofluorescence analysis of frozen embryonic brain sections was carried out as previously described29. Sagittal embryonic brain sections were incubated with anti-NES (1:200, Abcam) or anti-GFAP antibody (1:200, Millipore), followed by Alexa Fluor 555-conjugated goat anti-mouse IgG (1:200, Invitrogen) along with 0.1 μg/ml of DAPI. Apoptotic cells were detected by TUNEL assay using an ApopTag® red in situ apoptosis detection kit according to the manufacturer's protocol (Millipore).
Immunoblot analysis
For immunoblot analysis, cell lysates were prepared in hypotonic buffer and processed as previously described28. Briefly, total cell lysates (15 μg) were subjected to SDS-PAGE, followed by immunoblot detection with anti-TUJ1 (1:1,000, Millipore), anti-GFAP (1:1,000, Millipore), anti-NeuN (1:500, Millipore), anti-CASP3 (1:3,000, Cell Signaling), anti-Notch3 (1:200, Santa Cruz Biotechnology), or anti-β-actin antibody (1:10,000, Sigma-Aldrich). Based on the types of primary antibodies, the appropriate HRP-conjugated goat anti-mouse or anti-rabbit IgG (1:10,000, Enzo Life Sciences) or HRP-conjugated donkey anti-goat IgG (1:5,000, Santa Cruz Biotechnology) was used.
Quantitative real-time RT-PCR
Quantitative real-time RT-PCR (qRT-PCR) was carried out essentially as previously described with slight modifications28. Briefly, total RNA was isolated from cells using an RNeasy kit (Qiagen) and 10 ng of total RNA was used as a template for reverse transcription using the GoScript™ Reverse Transcription System (Promega). For qRT-PCR, we used an EvaGreen qPCR Mastermix-iCycler kit (Applied Biological Materials) and iCycler system (Bio-Rad). The mRNA expression levels of Gfap, Fox3, Nes, Notch1, Hes5, Hey1 and Neurog1 were normalized to the level of Gapdh. Primers used for qRT-PCR are as follows: Gfap-F (5′-CGA GTC CCT AGA GCG GCA AAT G-3′); Gfap-R (5′-GTA GGT GGC GAT CTC GAT GTC-3); Fox3-F (5′-CCA GGC ACT GAG GCC AGC ACA CAG C-3′); Fox3-R (5′-CTC CGT GGG GTC GGA AGG GTG G-3′); Nes-F (5′-GAA GCC CTG GAG CAG GAG AAG CA-3′); Nes-R (5′-TCC AGG TGT CTG CAA CCG AGA GTT C-3′); Notch1-F (5′- GTG TCG TGT GTC AAG CTG ATG-3′); Notch1-R (5′- CAT CCT GGG TTG TGC TCT TAG-3′); Hes5-F (5′-GCA GCA TAG AGC AGC TGA AG-3′); Hes5-R (5′-AGG CTT TGC TGT GTT TCA GG-3′); Hey1-F (5′-AAA ATG CTG CAC ACT GCA GG-3′); Hey1-R (5′-CGA GTC CTT CAA TGA TGC TCA G-3′); Neurog1-F (5′-ACC TGT CCA GCT TCC TCA CC-3′); Neurog1-R (5′-GTT CCT GCT CTT CGT CCT G-3′); Gapdh-F (5′-GGC ATT GCT CTC AAT GAC AA-3′); Gapdh-R (CTT GCT CAG TGT CCT TGC TG-3′).
Statistical analysis
Two-tailed unpaired Student's t-tests or two-way analysis of variance (ANOVA) with the Holm-Sidak method were used to compare the data between experimental and control groups. Although P < 0.05 was considered to be statistically significant in most cases, P < 0.1 was also noted in certain cases.
References
Hershko, A. & Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998).
Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405–439 (1996).
Ravid, T. & Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9, 679–690 (2008).
Wilkinson, K. D. et al. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246, 670–673 (1989).
Kawabe, H. & Brose, N. The role of ubiquitylation in nerve cell development. Nat. Rev. Neurosci. 12, 251–268 (2011).
Tai, H. C. & Schuman, E. M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838 (2008).
Ryu, K. Y. et al. Loss of polyubiquitin gene Ubb leads to metabolic and sleep abnormalities in mice. Neuropathol. Appl. Neurobiol. 36, 285–299 (2010).
Ryu, K. Y., Garza, J. C., Lu, X. Y., Barsh, G. S. & Kopito, R. R. Hypothalamic neurodegeneration and adult-onset obesity in mice lacking the Ubb polyubiquitin gene. Proc. Natl. Acad. Sci. USA 105, 4016–4021 (2008).
Davis, A. A. & Temple, S. A self-renewing multipotential stem cell in embryonic rat cerebral cortex. Nature 372, 263–266 (1994).
Gage, F. H. Mammalian neural stem cells. Science 287, 1433–1438 (2000).
McKay, R. Stem cells in the central nervous system. Science 276, 66–71 (1997).
Qian, X. et al. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28, 69–80 (2000).
Temple, S. The development of neural stem cells. Nature 414, 112–117 (2001).
Pierfelice, T., Alberi, L. & Gaiano, N. Notch in the vertebrate nervous system: an old dog with new tricks. Neuron 69, 840–855 (2011).
Yoon, K. & Gaiano, N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat. Neurosci. 8, 709–715 (2005).
Kageyama, R., Ohtsuka, T., Shimojo, H. & Imayoshi, I. Dynamic regulation of Notch signaling in neural progenitor cells. Curr. Opin. Cell Biol. 21, 733–740 (2009).
Kim, K. K., Adelstein, R. S. & Kawamoto, S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J. Biol. Chem. 284, 31052–31061 (2009).
Bilang-Bleuel, A. et al. Psychological stress increases histone H3 phosphorylation in adult dentate gyrus granule neurons: involvement in a glucocorticoid receptor-dependent behavioural response. Eur. J. Neurosci. 22, 1691–1700 (2005).
Prigent, C. & Dimitrov, S. Phosphorylation of serine 10 in histone H3, what for? J. Cell Sci. 116, 3677–3685 (2003).
Itoh, M. et al. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell 4, 67–82 (2003).
Yoon, K. J. et al. Mind bomb 1-expressing intermediate progenitors generate notch signaling to maintain radial glial cells. Neuron 58, 519–531 (2008).
Kageyama, R., Ohtsuka, T. & Kobayashi, T. Roles of Hes genes in neural development. Dev. Growth Differ. 50 Suppl 1, S97–103 (2008).
Ables, J. L., Breunig, J. J., Eisch, A. J. & Rakic, P. Not(ch) just development: Notch signalling in the adult brain. Nat. Rev. Neurosci. 12, 269–283 (2011).
Bi, F. et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. USA 110, 4069–4074 (2013).
Cahoy, J. D. et al. A transcriptome database for astrocytes, neurons and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 (2008).
Lee, S. et al. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J. Neurosci. 29, 234–249 (2009).
Park, H., Yoon, M. S. & Ryu, K. Y. Disruption of polyubiquitin gene Ubc leads to defective proliferation of hepatocytes and bipotent fetal liver epithelial progenitor cells. Biochem. Biophys. Res. Commun. 435, 434–440 (2013).
Ryu, K. Y. et al. The mouse polyubiquitin gene UbC is essential for fetal liver development, cell-cycle progression and stress tolerance. EMBO J. 26, 2693–2706 (2007).
Ryu, K. Y., Park, H., Rossi, D. J., Weissman, I. L. & Kopito, R. R. Perturbation of the hematopoietic system during embryonic liver development due to disruption of polyubiquitin gene Ubc in mice. PLoS ONE 7, e32956 (2012).
Shimojo, H., Ohtsuka, T. & Kageyama, R. Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron 58, 52–64 (2008).
Hirata, H. et al. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 298, 840–843 (2002).
Ryu, K. Y. et al. The mouse polyubiquitin gene Ubb is essential for meiotic progression. Mol. Cell. Biol. 28, 1136–1146 (2008).
Acknowledgements
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111485) to K.-Y.R.
Author information
Authors and Affiliations
Contributions
H.-W.R. and K.-Y.R. conceived and designed the research. H.-W.R. and C.-W.P. performed the experiments and analyzed the data. H.-W.R. and K.-Y.R. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Suppmentary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ryu, HW., Park, CW. & Ryu, KY. Disruption of polyubiquitin gene Ubb causes dysregulation of neural stem cell differentiation with premature gliogenesis. Sci Rep 4, 7026 (2014). https://doi.org/10.1038/srep07026
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07026
This article is cited by
-
Simultaneous Disruption of Both Polyubiquitin Genes Affects Proteasome Function and Decreases Cellular Proliferation
Cell Biochemistry and Biophysics (2020)
-
Ubiquitylation at the crossroads of development and disease
Nature Reviews Molecular Cell Biology (2018)
-
Temporal downregulation of the polyubiquitin gene Ubb affects neuronal differentiation, but not maturation, in cells cultured in vitro
Scientific Reports (2018)
-
Affinity of Tau antibodies for solubilized pathological Tau species but not their immunogen or insoluble Tau aggregates predicts in vivo and ex vivo efficacy
Molecular Neurodegeneration (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
