
Abstract
This study was aimed to determine the role and regulation of progranulin (PGRN) in the pathogenesis of inflammatory bowel diseases (IBD). Dextran sulfate sodium (DSS)−, picrylsulfonic acid (TNBS)-induced, bone marrow chimera and CD4+CD45Rbhi T cell transfer colitis model were established and analyzed in wild-type and several genetically-modified mice, including PGRN, IL-10 and TNFR2 deficient mice. Elevated levels of PGRN were found in colitis samples from human IBD patients and mouse colitis models in comparison to the corresponding controls. PGRN-deficient mice became highly susceptible to DSS- and TNBS-induced colitis, whereas recombinant PGRN ameliorated the pathology and reduced the histological score in both DSS and TNBS colitis models. In addition, hematopoietic-derived PGRN was critical for protection against DSS-induced colitis and lack of PGRN signaling in CD4+ T cells also exacerbated experimental colitis. PGRN-mediated protective effect in colitis was compromised in the absence of IL-10 signaling. In addition, PGRN's effect was also largely lost in the TNFR2-deficient colitis model. Collectively, these findings not only provide the new insight into PGRN's anti-inflammatory action in vivo, but may also present PGRN and its derivatives as novel biological agent for treating IBD.
Similar content being viewed by others


Introduction
Inflammatory bowel diseases (IBD) results from a continuum of complex interactions between a quart of host-derived and external elements1. IBD including both Crohn's disease (CD) and Ulcerative colitis (UC), is characterized by an imbalance in the mucosal intestinal immune system and a shift towards the proinflammatory side2. Nearly 4 million people worldwide are affected by either Ulcerative colitis or Crohn's disease3. It has been well established that TNFα plays a crucial role in the pathogenesis of IBD and TNF-blockers represent a well-accepted therapeutic option in the treatment of IBD4,5. Since the TNF blockade only show clinical remission in 50–70% of patients of IBD, alternative biological therapies can be an attractive candidate for further research6,7.
Progranulin (PGRN), also known as granulin epithelin precursor (GEP), PC-cell-derived growthfactor (PCDGF), proepithelin and acrogranin, is a 593-amino-acid secreted growth factor8,9. PGRN plays a critical role in a variety of physiologic and disease processes, including early embryogenesis10, wound healing11, inflammation12 and host defense13. PGRN also function as a neurotrophic factor14,15,16,17. PGRN exerts its therapeutic effect in inflammatory arthritis through binding to TNFR and DR3 and in turn disturbing TNFα/TNFR and TL1A/DR3 interactions18,19,20. PGRN−/− bone marrow-derived macrophages (BMDMs) reduced IL-10 production and increased amounts of pro-inflammatory cytokines13. PGRN also suppresses TNFα and IL-6 secretion from activated microglia21. Recombinant PGRN was able to block TNFα-induced respiratory robust from neutrophils12.
It was reported that PGRN antibodies occurred frequently in patients with CD and UC22. Cytotoxicity assays showed a proinflammatory effect of PGRN antibodies on human colon HT29 cells. Moreover, PGRN-antibodies, opposite to recombinant PGRN, enhanced TNF-α-induced downregulation of FOXP3 in CD4+CD25hi Tregs22. In this study, we investigated the role of PGRN in intestinal inflammation and the potential molecular mechanism involved.
Methods
Human biopsies
Collection of biopsies from human colons was carried out in accordance with the guidelines and approved by IRB and informed consent was obtained from all subjects during colonoscopy procedures at the Mount Sinai Hospital, New York in collaboration with Dr. Lloyd Mayer.
Mice
All of the animal studies were approved by the Institutional Animal Care and Use Committee of New York University (IACUC protocol #130202-01). Experiments were carried out in accordance with the approved guidelines and all used protocols approved by the committee's on animal care. Wild-type, TNFR2−/−, Rag1−/−, IL-10−/− mice were obtained from Jackson Laboratories (C57BL/6 background). The generation and characterization of PGRN knockout mice has been described previously13. All mice were maintained in accordance with the approved guidelines.
Induction of colitis
DSS colitis was induced by addition of 3% dextran sodium sulfate (DSS; 36,000–50,000 MW; MP Biomedicals) to the drinking water for 5 days, then replaced DSS solution with normal water23. The disease progression was determined by body weight changes, the presence of rectal bleeding and stool consistency. The bleeding score and stool score were evaluated according to the reference 23. Histology score of colonic tissue sections were scored in a blinded fashion as a combination of inflammatory cell inflitration (score 0–3) and tissue damage (score 0–3) according to the reference 24.
For TNBS model, mice were intrarectal instillation of 150 mg/kg picrylsulfonic acid (TNBS) dissolved in 100 μl 50% ethanol. Mice instilled 100 μl 50% ethanol as the control group25.
Bone marrow chimeras
6-weeks-old wild type mice (CD45.2) were lethally irradiated by two exposures of 550 rads (4 hour apart) prior to receiving 2 × 106 BM cells from wild type or PGRN-deficient mice (CD45.2) by intravenous injections. The radiation sensitivity of different cell types in bone marrow is distinct. Stem cells, T helper cells, cytotoxic T cells, monocytes and neutrophils are sensitive, while regulatory T cells, macrophages, dendritic cells and natural killer cells are radio-resistant26,27. To evaluate the radiation sensitivity and chimeric efficiency, we generated a control chimeras by reconstitution wild type recipients (CD45.2, three mice per group) with bone marrow cells from CD45.1 donors. In brief, bone marrows were collected from both the femurs and tibias of WT or PGRN-deficient donor mice by flushing with gauge needle and syringe. After passing cell suspension through 70 μM cell strainer and washing several times with PBS, cells were resuspended in PBS at a concentration of 2 × 107/ml. Injection of bone marrow cells should be done within 24 hours after irradiation. Anesthetize recipient mice with an Isoflurane vaporizer and inject 100 μl of cells suspension into the veinous network of eyeball. Monitor the reconstitute mice until they are fully awake. To avoid infection, we added sulfatrim to the drinking water (5 mL/200 mL) and maintain the mice on anti-biotic water for 2 weeks. 8 weeks after bone marrow transfer, radiation and chimeric efficiency was confirmed. For control chimera mice, FACS results show 96% of the transplanted CD45.1 bone marrow cells are present in reconstituted CD45.2 mice, suggesting a successful chimerism. To further verify the chimeric efficiency in WT/WT and WT/KO chimeras, we isolated and genotyped total bone marrow cells. WT mice with KO-BM gave rise to a 211-bp (−/−, PGRN-deficient alleles) PCR product, whereas WT mice with WT-BM produced a 468-bp PCR product (+/+, wild type alleles) in BM cells.
The WT/WT and WT/KO chimera mice were fed with 3% DSS for 5 days. Body weight changes, stool consistency and rectal bleeding were monitored daily. At day 7, mice were sacrificed to collect colon tissue for HE staining.
In vivo intestinal permeability assay
The intestinal barrier permeability was assessed with a FITC-labeled Dextran method as described28. In brief, mice were gavaged with FITC-dextran (Mw 4000; Sigma-Aldrich) at a concentration 60 mg/100 g body weight. FITC-dextran amount in serum was measured with a fluorescence spectrophotometer at emission and excitation wavelengths of 485 nm and 530 nm, respectively. FITC concentration was measured from standard curves generated by different dilution of FITC-dextran.
Assessment of microbiota
Microbial DNA was extracted with a QIAamp DNA stool kit (QIAGEN) according to the manufacture's protocols. The eluted DNA was qualified and qRT-PCR was performed with 16s rDNA primers. The forward and reverse primer sequences are as follows, 5′-GAGAGGAAGGTCCCCCAC-3′ and 5′-CGCTACTTGGCTGGTTCAG-3′ for bacteroides, 5′- ATGGCTGTCGTCAGCTCGT-3′ and 5′-CCTACTTCTTTTGCAACCCAC TC-3′ for enterbacteria, 5′-GCGGCGTGCCTAATACATGC-3′ and 5′-CTTCATCACTCA CGCGGCGT-3′ for bacillus, 5′-GCTGCTAATACCGCATGATATGTC-3′ and 5′-CAGACG CGAGTCCATCTCAGA-3′ for firmicutes. The data was presented as a percentage relative to eubacteria.
Establishment of CD4+CD45Rbhi T cell transfer colitis model
CD4+CD45Rbhi T cells were purified from the spleen cells of wild-type and PGRN-deficient mice through FACS sorting. The purity of the transferred CD4+CD45Rbhigh T cells was 98%. 4 × 105 syngeneic CD4+CD45Rbhigh T cells were injected i.v. into RAG1−/− mice to establish the transfer colitis model. The level of severity of colitis was assessed through evaluating body weight changes and histological scores.
Immunohistochemistry
Colonic tissues were fixed in 10% formalin, dehydrated, cleared with dimethylbenzene and then embedded in parafin. These samples were then incubated with anti-PGRN antibody (1:100 dilutions; Santa Cruz Biotechnology) at 4 degree overnight, followed by incubation with a horseradish peroxidase-conjugated secondary antibody for 60 min at room temperature. The signal was detected using the Vector Elite ABC Kit (Vectastain; Vector). These sections were also stained using hematoxylin and eosin (HE) for routine morphologic analysis.
Immunoblotting
The human intestinal epithelial Caco2 cells were lysed in RIPA lysis buffer containing protease inhibitors. Proteins were resolved on a 10% SDS-polyacrylamide gel and electroblotted onto a nitrocellulose membrane. After blocking in 5% non-fat dry milk in Tris buffer-saline-Tween 20 (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; and 0.5% Tween 20), blots were incubated with with anti-Erk, -Phospho-Erk (Cell Signaling Techonology) antibody for 1 hour. After washing, the secondary antibody (horseradish peroxidaseconjugated anti-rabbit immunoglobulin; 1:2000 dilution) was added and bound antibody was visualized using an enhanced chemiluminescence system (Amersham Life Science, Arlington Heights, IL, USA).
Flow cytometry
Cells were stained with anti-CD16/32,-CD3, -CD4, -CD8, -CD11b, -CD11c, -CD19, -CD25, -MHCII, -mPDCA-1 (eBioscience). Intracellular staining for Foxp3 (eBioscience staining buffer set) and IL-17 and IFN-γ were conducted according to manufacturers' instructions (Fixation and Fixation and Permeabilization Solution Kit Biolegend and eBioscience, respectively). Data were acquired on a LSRII (BD Biosciences) and analyzed with FlowJo software (Tree star, Ashland OR).
Statistical analysis
Results represent mean ± SEM. Differences in groups were analyzed with unpaired, 2-tailed t tests. P values less than 0.05 were considered significant.
Results
PGRN is elevated in the colon from IBD patients and mice colitis model
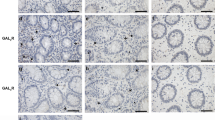
To determine the expression of PGRN in intestinal tissue during inflammatory conditions we performed immunohistochemistry in the human intestinal tissue samples. As shown in Fig. 1A, the expression of PGRN is significantly elevated in the intestinal tissue of IBD patients compared with noninflammatory tissue controls. PGRN was detectable in normal colon tissues (day 0) and its expression was significantly increased at day 1 after TNBS injection (Fig. 1B). Stronger PGRN staining was also observed in epithelial cells and inflammatory cell infiltrating areas at day 4 (Fig. 1B). We performed real-time PCR to examine PGRN gene expression pattern in immune cells sorted from normal wild-type mice spleens. We found that PGRN gene was highly expressed in CD11b+ cells and Ly6G+ cells compared to other immune cells including T and B cells (Fig. 1C). To determine whether PGRN is functional on the intestinal cells, we stimulated the human intestinal epithelial Caco2 cells with 1 μg/ml recombinant PGRN, we found that PGRN activated Erk phosphorylation (Fig. 1D). These data suggests that PGRN is induced in the IBD inflammatory conditions and is functional on the intestinal cells.

PGRN expression is increased in intestinal samples from human IBD patients and mouse colitis models.
(A) Immunohistochemistry for PGRN was conducted on sections of noninflammatory bowel disease and IBD patient's intestinal tissue. (B) PGRN expression was detected in colonic sections from Babl/c mice after TNBS induction at different time-points. (C) Real-time PCR analysis for PGRN gene expression in different cells sorted from wild-type (C57BL/6) spleens. (D) Human intestinal Caco2 cells were treated with PGRN at different time-points and the Erk signaling was examined.
PGRN deficient mice develop more severe colitis in comparison to the wild-type mice in chemical-induced colitis model
To study the contribution of endogenous PGRN to colitis development, we assessed the age- and sex-matched wild-type and PGRN−/− mice after oral administration of 3% DSS in drinking water. In a survival study, no wild-type mice died until day 10 after DSS induction, but a mortality higher than 60% was observed in the PGRN−/− group (Fig. 2A). The PGRN−/− mice suffered from significant body weight loss from day 4 (day 4: 97.8 ± 1.2% vs. 91.6 ± 5.2%, p = 0.02; day 5: 96.1 ± 0.9% vs. 87.3 ± 4.7%, p = 0.002; day 6: 90.3 ± 1.5% vs. 79.5 ± 4.6%, p = 0.0008; day 7: 86.3 ± 1.7% vs. 75.6 ± 2.2%, p = 0.0005) (Fig. 2B). In addition, the stool consistency scores of PGRN−/− mice became significantly worse compared to those of DSS-fed wild-type mice (day 5, p = 0.005; day 6, p = 0.04; day 7, p = 0.005) (Fig. 2C). And PGRN−/− mice displaying significantly elevated bleeding scores (day 4, p = 0.03; day 5, p = 0.005; day 6, p = 0.013; day 7, p = 0.009) (Fig. 2D) relative to DSS-induced wild-type mice. The evaluation of colon length is one of the parameter with the lowest variability in DSS-induced colitis model model29. We found that the colons of PGRN−/− mice were on average 20% shorter than those of wild-type mice treated with DSS (7.5 ± 0.5 vs. 6.5 ± 0.5, p = 0.03) (Fig. 2E). Colitis tissue from DSS-administered mice were examined to determine whether clinical signs of colitis were correlated with histological severity. Marked histopathological changes were seen in colonic sections of DSS-fed PGRN−/− mice characterized by severe transmural inflammation with focal areas of extensive ulceration and necrotic lesions, along with infiltration of inflammatory cells (Fig. 2F). Semiquantitative scoring of these histological parameters confirmed that colitis severity in PGRN−/− mice was obviously higher than that in wild-type mice (p < 0.01) (Fig. 2G).

PGRN−/− mice are highly susceptible to DSS induction.
DSS colitis model was established in wild-type (n = 10) and PGRN−/− (n = 8) mice by administration of 3% DSS solution in drinking water for 5 days, followed by normal drinking water for 2 days. (A) Survival rate was monitored until day 13 after DSS induction. (B) Body weight. (C) Stool consistency. (D) Bleeding scores. (E) Mice were sacrificed on day 7 to measure colon length. (F) Histopathological changes. (G) Semiquantitative scoring of histopathology was performed as described in Methods. Scale bars, 200 μm.
To determine whether these observations were specific to DSS-induced colitis model, another acute colitis model which resembles Crohn's disease was established through intrarectal administration of 150 mg/kg picrylsulfonic acid (TNBS) in ethanol. Compared with WT mice, PGRN−/− mice significantly lost more body weight at day 3 and 4 after TNBS induction (day 3: 88.7 ± 4.6% vs. 82.3 ± 4.6%, p = 0.039; day 4: 94.7 ± 3.8% vs. 86.1 ± 2.3%, p = 0.005) (Fig. 3A). In addition, the histological features of PGRN−/− mice were significantly different from wild-type mice (Fig. 3B). Taken together, these results suggest that PGRN- dependent signaling is critical for protection against DSS- and TNBS-induced acute colitis.

PGRN−/− mice are highly susceptible to TNBS-induced colitis.
(A) Body weight of WT (n = 6) and PGRN−/− mice (n = 5). (B) HE staining of representative colonic sections. Scale bars, 400 μm.
Hematopoietic-derived PGRN is critical for protection against DSS-induced colitis
PGRN is highly expressed in epithelial cells as well as in immune cells19. To investigate whether immune cell-derived PGRN plays a role in DSS-induced colitis model, we generated chimera mice made by reconstituting irradiated wild-type mice with bone marrow cells from KO/WT donors, then subjected them to 3% DSS solution for 5 days. The wild-type mice reconstituted with PGRN−/− bone marrow cells lost more body weight compared to wild-type mice transplanted with wild-type bone marrow (day 4: 101.4 ± 2.1% vs. 98.8 ± 2.2%, p = 0.03; day 5: 100.5 ± 0.8% vs. 98.4 ± 2.9%, p = 0.05; day 6: 99.5 ± 0.7% vs. 96.5 ± 3.1%, p = 0.019; day 7: 97.3 ± 1.1% vs. 94.6 ± 1.8%, p = 0.011) (Fig. 4A). HE stained colon sections of wild-type mice that received PGRN−/− bone marrow displayed severe epithelial ulceration and more inflammatory lymphocytes infiltration (Fig. 4B) and significantly higher scores (p < 0.01, Fig. 4C). We found that wild-type mice transplanted with wild-type bone marrow were less sensitive to DSS-induced colitis. Collectively, these results demonstrate that hematopoietic cell-derived PGRN protects against DSS-induced colitis.

PGRN signaling in hematopoietic cells is important for protection against DSS-induced colitis.
WT/WT (n = 6) and WT/PGRN−/− (n = 6) mice were fed a 3% DSS solution in drinking water for 5 days, followed by normal drinking water for 2 days. (A) Body weight, (B) Histopathological changes in colon tissue. (C) Semiquantitative scoring of histopathology. Scale bars, 200 μm.
Lack of PGRN signaling in CD4+ T cells also exacerbates experimental colitis
To determine whether PGRN played a role in chronic colitis model, we established CD4+CD45Rbhigh T cells transfer colitis model. The results demonstrated that transfer of PGRN−/− CD4+CD45Rbhigh T cells led to an accelerated onset of body weight loss at d28 and d34 (p < 0.01) (Fig. 5A). RAG1−/− mice that received PGRN−/− T cells had a greater rate of mortality (Fig. 5B) and showed marked alterations of the colon (Fig. 5C). In line with the findings of accelerated body weight loss, lack of PGRN signaling in CD4+ T cells leads to more severe signs of intestinal inflammation.

Lack of PGRN signaling in CD4+ T cells execrates experimental colitis.
(A) Body weight following adoptive transfer of CD4+CD25-CD45Rbhi T cells from WT and PGRN−/− donors into Rag1−/− recipients. (B) Survival rate of Rag1−/− mice reconstituted with WT and PGRN−/− CD4+CD25-CD45Rbhi T cells. (C) Representative colonic sections from Rag1−/− mice reconstituted with WT and PGRN−/− CD4+CD25-CD45Rbhi T cells. All data represent means ± SE of a representative experiment. *p < 0.05; **p < 0.01. Scale bars, 200 μm.
The severity of colonic inflammation is not resulted from the impairment of intestinal barrier function or the alternations of colonic microbiota in DSS-fed PGRN−/− mice
Impairment of the intestinal barrier further drives the pathogenesis of intestinal inflammation in DSS-induced colitis and human IBD30. The results revealed that no significant changes were observed between wild-type and PGRN−/− DSS mice (water: 19.3 ± 3.0 vs. 18.2 ± 5.7, p = 0.39; DSS model: 131.5 ± 25 vs. 131.8 ± 20, p = 0.495) (Fig. 6A). These results suggest that PGRN deficiency does not affect the function of the intestinal barrier at either physiologic or disease conditions.

Intestinal barrier function and the microbiota are not altered in PGRN−/− mice.
(A) Intestinal permeability was examined by detection of a FITC-dextran in the serum from DSS-induced mice. (B–E) Q-PCR analysis of intestinal microbiota in naïve or DSS-treated wild-type and PGRN−/− mice. Values are shown as a relative ratio to total bacterial 16 s rDNA measured by 2−ΔΔct method. All data represent means ± SE of three independent experiments. *p < 0.05; **p < 0.01.
Intestinal microbiota plays a crucial role in the pathogenesis of inflammatory bowel disease31. To determine whether lack of PGRN signaling affects intestinal bacterial overgrowth and leads to bacteremia, we performed bacteria assay. Unexpectedly, we did not observe any significant changes in the number of bacteria between naïve and DSS-treated wild-type and PGRN−/− mice (Fig. 6 B–E). These results demonstrate that the severity of colitis in PGRN−/− mice is not caused by bacterial overgrowth or due to the alteration of the composition of intestinal microbiota.
Recombinant PGRN ameliorates colitis syndrome
In order to determine whether recombinant PGRN is able to ameliorate the colitis syndrome in wild-type mice, a therapeutic group of mice (n = 8) were treated with 100 μg PGRN every two days beginning at day 1 after DSS induction, whereas control group (n = 8) were treated with PBS. The PBS group exhibited the great amounts of body weight loss and were unable to recover as rapidly in comparison to the PGRN-treated group (day 6: 93 ± 3.7% vs. 98 ± 3.5%, p = 0.02; day 7: 90 ± 5.0% vs. 95 ± 3.2%, p = 0.02; day 8: 88.6 ± 6.7% vs. 96.8 ± 4.7%, p = 0.009) (Fig. 7A). And the colon was shorter in control group than in mice treated with recombinant PGRN (p < 0.05) (Fig. 7B). PGRN treatment significantly increased the IL-10 release in colonic explants from DSS colitis mice (p < 0.01) (Fig. 7C). Histological results also showed that recombinant PGRN protected against experimentally induced colonic hyperplasia and leukocyte infiltration in colonic tissues (Fig. 7D).

Recombinant PGRN attenuates the inflammation in DSS-induced colitic mice.
(A) Body weight of PBS treated versus PGRN treated DSS induced colitis mice. Per group 8 mice were used. (B) Representative macroscopic pictures of colonic sections from mice treated with PBS or PGRN. (C) Levels of IL-10 were examined after colonic tissue explants were harvested at d8 and cultured ex vivo at 37°C for 24 h. (D) Representative HE-stained colon sections from mice treated with PBS and PGRN. All data represent means ± SE of three independent experiments. *p < 0.05; **p < 0.01. Scale bars, 200 μm.
DSS-induced colitis was also observed in immunodeficient mice32. In a separate group, we treated colitic Rag1−/− mice with PBS and PGRN as described above. Colitic Rag1−/− mice treated with recombinant PGRN lost significantly less body weight when compared with those receiving PBS (day 6: 82 ± 8.7% vs. 91 ± 5.9%, p = 0.04; day 7: 81.1 ± 9.7% vs. 91.4 ± 4.5%, p = 0.02) (Fig. 8A). Colons of PBS-treated DSS-fed mice were significantly shorter than those of recombinant PGRN-treated colitic mice (Fig. 8B). Furthermore, HE staining of colonic sections confirmed the amelioration in colitis severity in PGRN-treated colitic mice characterized by focal areas of reduced ulceration and less leukocyte infiltration (Fig. 8C).

Recombinant PGRN attenuates the severity of DSS-induced colitis established in Rag1−/− mice.
(A) Body weight. Rag1−/− mice were subjected to DSS-induced colitis and their body weight was measured. 5 mice per group were used. (B) Statistics analysis of colon length of each group. (C) Representative HE-stained colon sections of mice treated with PBS and PGRN. All data represent means ± SE of three independent experiments. *p < 0.05; **p < 0.01. Scale bars, 200 μm.
IL-10 signaling is required for PGRN-mediated protection from intestinal inflammation
IL-10 plays a critical role in regulating inflammatory response in intestine since the most prominent phenotype observed in IL-10-deficient mice is inflammation in the intestine33. PGRN−/− mice macrophages produce less IL-10 than wild-type mice and the serum IL-10 level is significantly elevated in PGRN-treated inflammatory arthritis model18. To determine whether IL-10 signaling is required for PGRN-mediated protection from colitis, we blocked the IL-10 signaling with a specific anti-IL-10 receptor antibody (clone 1B1.3A). Control group mice (n = 5) were injected with recombinant PGRN and isotype antibody, whereas blocking group mice (n = 5) received PGRN and 200 μg of anti-IL10R mAb per mice every 2 days. As shown in Fig. 9A, the anti-IL10R mAb blocking group mice suffered from significant body weight loss from day 4 (day 4: 97.8 ± 2.2% vs. 91.6 ± 2.0%, p = 0.001; day 5: 99.1 ± 1.1% vs. 93.8 ± 1.6%, p = 0.0002; day 6: 98.8 ± 3.5% vs. 90.4 ± 0.6%, p = 0.007; day 7: 95.4 ± 4.7% vs. 86.9 ± 5.2%, p = 0.013; day 8: 97.4 ± 6.1% vs. 85 ± 6.6%, p = 0.007; day 9: 98.6 ± 6.8% vs. 84.2 ± 6.9%, p = 0.005). These results demonstrated that anti-10R mAb offsets the protection of PGRN against body weight loss. Marked inflammatory cell infiltration, colonic ucleration and changes of crypt architecture were also observed in intestinal sections of anti-10R treatment mice (Fig. 9B). In addition, PGRN's therapeutic effect was lost in IL-10−/− mice, at least in part (day 3: 87.1 ± 2,5% vs. 84.4 ± 5.1%, p = 0.16; day 4: 86.7 ± 2.2% vs. 88.3 ± 4.8%, p = 0.26; day 5: 88.2 ± 3.3% vs. 90.5 ± 4.2%, p = 0.20) (Fig. 9C). Intestinal sections of the PGRN group and PBS group have similar histological changes (Fig. 9D). No significant changes were observed in Foxp3, IFN-γ and IL-17 level in T cells isolated from MLN between PGRN and PBS group (CD4+IL-17+ cells: 2.52 ± 0.5% vs. 2.07 ± 0.8%, p = 0.21; CD4+IFN-γ+ cells: 1.51 ± 0.48% vs. 1.62 ± 0.69%, p = 0.21; CD4+Foxp3+ cells: 10.5 ± 1.7% vs. 9.88 ± 2.1%, p = 0.49) (Fig. 9E). These results suggest that IL-10 signaling is required for PGRN-mediated protection from intestinal inflammation.

IL-10 signaling is required for PGRN-mediated protection from experimental colitis.
(A) DSS colitis was established in wild-type mice, 5 mice were injected with an IL-10R monoclonal antibody to block IL-10 signaling whereas 5 mice were injected with an isotype antibody, then body weight was measured. (B) Representative intestinal sections from mice treated with isotype antibody or anti-IL10R mAb. (C) Body weight. TNBS colitis was established in IL-10−/− mice and these mice were treated with PBS or PGRN. Eight mice per group were used. (D) Representative histological changes of colon sections from IL-10−/− mice treated with PBS or PGRN. (E) Foxp3, IFN-γ and IL-17 level of T cells from MLN were detected by FACS method. All data represent means ± SE of three independent experiments. *p < 0.05; **p < 0.01. Scale bars, 200 μm.
The protective effect of PGRN in intestinal inflammation depends on TNFR2 as well
Several publications have demonstrated that TNFR2 is important for PGRN-mediated protective roles18,34,35,36,37. To define the potential contributions of TNFR2 in the therapeutic effect of PGRN, we established TNBS model in TNFR2−/− mice and treated with PBS or 100 μg PGRN every two days beginning at day 1 after TNBS induction. The results revealed that no significant difference in body weight changes (Fig. 10A) and histological features of colonic sections (Fig. 10B) between PBS- and PGRN-treated colitic TNFR2−/− mice (n = 5, p > 0.05). Collectively, this set of experiments indicated that TNFR2−/− colitic mice are less sensitive to PGRN treatment and PGRN-mediated protection against intestinal inflammation may also rely on TNFR2 signaling, at least in part.

PGRN loses protective effect on intestinal inflammation in TNFR2-deficient colitis model.
TNFR2−/− TNBS colitis model was induced by administration of TNBS in ethanol. PGRN group mice (n = 5) were treated with 100 μg PGRN every two days beginning at day 1 after TNBS induction, whereas control group (n = 5) were treated with PBS. (A) Body weight, (B) Histopathological changes in colon tissue were examined by H&E staining. All data represent means ± SE of three independent experiments. *p < 0.05; **p < 0.01. Scale bars, 200 μm.
Discussion
The objective of this study was to examine the potential role of PGRN in the pathogenesis of IBD as well as the molecular mechanism involved. It was reported that PGRN expression was increased during colonic inflammation and PGRN was a critical protein involved in colonic injury repair38. We further revealed that PGRN expression was elevated in the patients with IBD and its level was induced in the course of mouse experimental colitis (Fig. 1), suggesting that PGRN might be an important growth factor involved in mucosal inflammatory response and damage. Although PGRN−/− mice have no evidence of immune system activation and spontaneous colitis phenotype (Fig. 11), our findings demonstrate that PGRN deficiency exacerbates the severity of acute mucosal inflammation induced by chemical agents. PGRN−/− mice were significantly more susceptible to colitis-associated body weight loss, diarrhea, mortality, colon length and histological changes (Figs. 2, 3), indicating a critical role for the PGRN in protection against DSS- and TNBS-induced colitis. In addition, recombinant PGRN significantly reduced the severity of DSS-induced colonic injury assessed by clinical, biological and histological parameters(Fig. 7).

PGRN−/− mice have normal immune cells numbers and do not develop spontaneous colitis.
(A) DC cells from the spleen of wild-type and PGRN−/− mice were gated with CD11c and MHCII and then further examined with CD11b and CD8 markers using flow cytometry. (B) Spleen and MLN cells from WT and KO mice were assessed for CD4+Foxp3+ T cells. (C) Splenic plasmacytoid DC (mPDCA-1+CD3−CD19−) were assessed by flow cytometry. (D) Histopathological changes in colon tissue from WT and KO mice were examined by H&E staining. (n = 3 mice/genotype, mean ± SD, *p < 0.05). Scale bars, 200 μm.
Bone marrow chimera mice results demonstrate that hematopoietic cell-derived PGRN was sufficient for protection against colitis (Fig. 4). PGRN also exerts its beneficial effect even in the absence of both T and B cells (Fig. 8). However, this finding does not belittle the role of T cells- and B cells-derived PGRN in chronic DSS colitis and other IBD models. Several recent articles have indicated that PGRN contributes to the pathogenesis of chronic autoimmune diseases19,39,40. In CD4+CD45Rbhigh T cells transfer colitis model, transfer of PGRN−/− CD4+CD45Rbhigh T cells leads to more severe intestinal inflammation (Fig. 5). Intriguingly, PGRN is highly expressed in CD11b+ cells and Ly6G+ cells (Fig. 1C) and adoptive transfer of CD11b+Ly6G+ myeloid-derived suppressor cells (MDSCs) has been reported to decrease intestinal inflammation41. Studies are warranted to elucidate the role of PGRN expressed in CD11b+ and Ly6G+ cells in the pathogenesis of colitis.
PGRN binds to TNFR2 with higher binding affinity18,42 and TNFR2 has been shown to be important for PGRN activities36. TNFR2 has also been found to be required for PGRN-mediated protection of LPS-induced lung injury34. In addition, PGRN-stimulated bone formation and fracture healing also depended on TNFR2 signaling35. Our finding that PGRN lost its therapeutic effect, at least in part, in TNBS model established in TNFR2-deficient mice (Fig. 10) corresponds with previous reports and supports the concept that TNFR2 plays an important role in various PGRN-mediated pathophysiological conditions. Interestingly, a lack of TNFR2 expression within CD4+ T cells was also reported to exacerbate the development of T cells transfer colitis in mice43. It is noted that Chen and colleagues agreed that PGRN stimulation of regulatory T cells (Treg) was mediated by TNFR2. They reported that PGRN stimulates mouse Tregs through enhancing TNFα-induced Treg36. The effect of TNFα on Tregs from mice and humans remains to be controversial. The data from Chen lab suggest that TNFα promotes murine Treg in vitro36, whereas in humans, TNFα inhibits the suppressive function of Tregs through negative regulation of Foxp3 expression21,44,45,46,47. Thus, it appears that PGRN stimulates the suppressive function of both human and mouse Tregs, although the exact molecular mechanism remains to be further delineated. Although the effect of TNFα on Treg function remains controversy, the beneficial and therapeutic effects of Tregs in autoimmune diseases have been well-accepted48,49,50. In addition, TNF inhibitors, including Enbrel, Remicade, Humira, have been accepted as the most effective anti-inflammatory therapeutics and the most successful biotech drugs51,52. Importantly, recent studies reported that PGRN antibodies occurred frequently in patients with both CD and UC and had significant neutralizing effects on PGRN plasma levels22. These studies suggest that PGRN and its derivatives may be more effective alternatives to market TNF inhibitors for treating the IBD patients who are diagnosed to be PGRN antibodies positive.
The spontaneous colitis in IL-10R−/− mice and Blimp−/− mice, which exhibit a defect in IL-10 signaling53,54, suggests the importance of IL-10 in preventing intestinal inflammation. IL-10 has been identified as a potential therapeutic molecule in Crohn's disease and ulcerative colitis1,55,56. In line with the report that PGRN-treated mice showed a significant increase in IL-10 production in collagen-induced arthritis model18, we found that PGRN treatment also increased the levels of IL-10 in colitis models and PGRN largely lost its protective effect in colitis when IL-10 signaling was blunted by IL-10R blocking antibody or in IL-10-deficient mice. This further confirms that upregulation of IL-10 production is one of the mechanism involved in the anti-inflammatory effects of PGRN in various inflammatory conditions. To better understand the roles of TNFR2 signaling and IL-10 in PGRN-mediated anti-inflammatory effect, we proposed a model illustrated in Fig 12. In brief, PGRN binds to TNFR2 in target cells, exemplified with Tregs, leading to the secretion of IL-10, an anti-inflammatory cytokine known to be involved in various inflammatory diseases, including IBD, whereas TNFα may neutralize the anti-inflammatory effects of PGRN by interfering the PGRN/TNFR2 signaling.

A proposed model for explaining the involvements of TNFR2 signaling and Il-10 in PGRN-mediated anti-inflammatory activity in the target cells, exemplified with regulatory T cells.
Collectively, this study shows that PGRN is a critical mediator of intestinal homeostasis and its dysregulation associates with intestinal inflammation during colitis. In addition, the PGRN-mediated protective action in inflammatory bowel diseases depends on the TNFR2 and IL-10 signaling. These findings not only provide the new insights into PGRN's anti-inflammatory action in vivo, but may also present PGRN and its derivatives as novel strategy in the treatment of intestinal inflammation.
References
Kaser, A., Zeissig, S. & Blumberg, R. S. Inflammatory bowel disease. Annu Rev Immunol 28, 573–621 (2010).
Atreya, I., Atreya, R. & Neurath, M. F. NF-kappaB in inflammatory bowel disease. J Intern Med 263, 591–596 (2008).
Park, K. T. & Bass, D. Inflammatory bowel disease-attributable costs and cost-effective strategies in the United States: a review. Inflamm Bowel Dis 17, 1603–1609 (2011).
Fu, X. Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res 16, 214–219 (2006).
Scharl, M. & Rogler, G. [Biologicals in gastroenterology: TNF-blockers]. Dtsch Med Wochenschr 135, 2243–2252 (2010).
McNamee, E. N. et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci U S A 108, 16711–16716 (2011).
Targan, S. R. et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med 337, 1029–1035 (1997).
Anakwe, O. O. & Gerton, G. L. Acrosome biogenesis begins during meiosis: evidence from the synthesis and distribution of an acrosomal glycoprotein, acrogranin, during guinea pig spermatogenesis. Biol Reprod 42, 317–328 (1990).
Ong, C. H. & Bateman, A. Progranulin (granulin-epithelin precursor, PC-cell derived growth factor, acrogranin) in proliferation and tumorigenesis. Histol Histopathol 18, 1275–1288 (2003).
Sun, X., Gulyas, M. & Hjerpe, A. Mesothelial differentiation as reflected by differential gene expression. Am J Respir Cell Mol Biol 30, 510–518 (2004).
He, Z., Ong, C. H., Halper, J. & Bateman, A. Progranulin is a mediator of the wound response. Nat Med 9, 225–229 (2003).
Zhu, J. C. et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell 111, 867–878 (2002).
Yin, F. et al. Exaggerated inflammation, impaired host defense and neuropathology in progranulin-deficient mice. J Exp Med 207, 117–128 (2010).
Baker, M. I. R. et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919 (2006).
Cruts, M. I. et al. Null mutations in progranulin cause ubiquitin- positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924 (2006).
Gass, J. A. et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 15, 2988–3001 (2006).
Rowland, L. P. Frontotemporal dementia, chromosome 17 and progranulin. Ann Neurol 60, 275–277 (2006).
Tang, W. et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science 332, 478–484 (2011).
Jian, J. Konopka, J. & Liu, C. Insights into the role of progranulin in immunity, infection and inflammation. J Leukoc Biol 93, 199–208 (2013).
Liu, C. et al. Progranulin-derived Asttrin directly binds to TNFRSF25 (DR3) and inhibits TNF-like ligand 1A (TL1A) activity. PLOS ONE 9, e92743 (2014).
Pickford, F. J. et al. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am J Pathol 178, 284–295 (2011).
Thurner, L. et al. Proinflammatory Progranulin Antibodies in Inflammatory Bowel Diseases. Dig Dis Sci 59, 1733–1742 (2014).
Wirtz, S., Neufert, C., Weigmann, B. & Neurath, M. F. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2, 541–546 (2007).
Zaki, M. H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 et al. (2010).
Scheiffele, F. & Fuss, I. J. Induction of TNBS colitis in mice. Curr Protoc Immunol Chapter 15, Unit 15 19 (2002).
Green, D. E. et al. Consequences of irradiation on bone and marrow phenotypes and its relation to disruption of hematopoietic precursors. Bone 63, 87–94 (2014).
Heylmann, D. et al. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim Biophys Acta 1846, 121–129 (2014).
Ibla, J. C. & Khoury, J. Methods to assess tissue permeability. Methods Mol Biol 341, 111–117 (2006).
Okayasu, I., Hatakeyama, S., Yamada, M., Ohkusa, T., Inagaki, Y. & Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702 (1990).
Munkholm, P. et al. Intestinal permeability in patients with Crohn's disease and ulcerative colitis and their first degree relatives. Gut 35, 68–72 (1994).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9, 313–323 (2009).
Dieleman, L. A. et al. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107, 1643–1652 (1994).
Kamanaka, M. et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25, 941–952 (2006).
Guo, Z., Li, Q., Han, Y., Liang, Y., Xu, Z. & Ren, T. Prevention of LPS-induced acute lung injury in mice by progranulin. Mediators Inflamm 2012, 540794 (2012).
Zhao, Y. P., Tian, Q. Y., Frenkel, S. & Liu, C. J. The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials 34, 6412–6421 (2013).
Hu, Y., Xiao, H., Shi, T., Oppenheim, J. J. & Chen, X. Progranulin promotes TNF-induced proliferation of suppressive mouse CD4 Foxp3 regulatory T cells. Immunology 142, 193–201 (2014).
Zhao, Y. P. et al. Progranulin protects against osteoarthritis through interacting with TNF-α and β-catenin signaling. Annals of rheumatic diseases 2014-205779 (2014).
Reardon, C. et al. Thymic stromal lymphopoetin-induced expression of the endogenous inhibitory enzyme SLPI mediates recovery from colonic inflammation. Immunity 35, 223–235 (2011).
Thurner, L. et al. Progranulin antibodies in autoimmune diseases. J Autoimmun 42, 29–38 (2013).
Tanaka, A. et al. Serum progranulin levels are elevated in patients with systemic lupus erythematosus, reflecting disease activity. Arthritis Res Ther 14, R244 (2012).
Guan, Q. et al. The role and potential therapeutic application of myeloid-derived suppressor cells in TNBS-induced colitis. J Leukoc Biol 94, 803–11 (2013).
Jian, J. et al. Progranulin directly binds to the CRD2 and CRD3 of TNFR extracellular domains. FEBS Lett 587, 3428–3436 (2013).
Dayer Schneider, J. et al. Lack of TNFR2 expression by CD4(+) T cells exacerbates experimental colitis. Eur J Immunol 39, 1743–1753 (2009).
Ehrenstein, M. R. et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med 200, 277–285 (2004).
Nie, H. et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-alpha in rheumatoid arthritis. Nat Med 19, 322–328 (2013).
Valencia, X., Stephens, G., Goldbach-Mansky, R., Wilson, M., Shevach, E. M. & Lipsky, P. E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 108, 253–261 (2006).
Zanin-Zhorov, A. et al. Protein kinase C-theta mediates negative feedback on regulatory T cell function. Science 328, 372–376 (2010).
Chen, Z. et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 39, 272–285 (2013).
Komatsu, N. et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 20, 62–68 (2014).
Fantini, M. C. et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology 136, 1308–1316 (2009).
Charo, I. F. & Taub, R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov 10, 365–376 (2011).
Palladino, M. A., Bahjat, F. R., Theodorakis, E. A. & Moldawer, L. L. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov 2, 736–746 (2003).
Spencer, S. D. et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med 187, 571–8 (1998).
Martins, G. A. et al. Transcriptional repressor Blimp-1 regulates T cell homeostasis and function. Nat Immunol 7, 457–65 (2006).
Steidler, L. et al. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 289, 1352–1355 (2000).
Lindsay, J. O., Sandison, A., Cohen, P., Brennan, F. M. & Hodgson, H. J. IL-10 gene therapy is therapeutic for dextran sodium sulfate-induced murine colitis. Dig Dis Sci 49, 1327–1334 (2004).
Acknowledgements
We thank Dr. Lloyd Mayers who passed away on September 5, 2013 for the collaboration and support in several ways, including providing the human IBD samples and insightful discussions. We also thank Huizhong Xiong, Benny Yang and Jonathan Weiss for helpful discussion. This work was supported partly by NIH research grants R56AI100901, R01AR062207, R01AR061484 and a Disease Targeted Research Grant from Rheumatology Research Foundation.
Author information
Authors and Affiliations
Contributions
F.H.W. designed and performed experiments and wrote the paper. Y.Y.Z. performed experiments, acquired data and drafted the manuscripts. J.J.L. and J.J.M. performed immunohistochemistry assays. Q.Y.T. collected and analyzed data. J.Q.L. assisted in construction of bone marrow chimera model. J.J.L., W.T., W.M.Z. and X.P.Y. assisted in analyzing the data and editing the manuscript. C.J.L. supervised this study, analyzed data and edited the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wei, F., Zhang, Y., Jian, J. et al. PGRN protects against colitis progression in mice in an IL-10 and TNFR2 dependent manner. Sci Rep 4, 7023 (2014). https://doi.org/10.1038/srep07023
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07023
This article is cited by
-
Penfluridol targets acid sphingomyelinase to inhibit TNF signaling and is therapeutic against inflammatory autoimmune diseases
Arthritis Research & Therapy (2022)
-
Progranulin regulates the development and function of NKT2 cells through EZH2 and PLZF
Cell Death & Differentiation (2022)
-
Serum progranulin as a potential biomarker for frailty in Chinese older adults
Aging Clinical and Experimental Research (2022)
-
A novel mechanism of EAE resistance highlights the conflicting roles of progranulin-mediated immunosuppression and antigen processing
Cellular & Molecular Immunology (2021)
-
Progranulin deficiency exacerbates spinal cord injury by promoting neuroinflammation and cell apoptosis in mice
Journal of Neuroinflammation (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
