
Abstract
Autosomal dominant optic atrophy (ADOA) is the most frequent form of hereditary optic neuropathy and occurs due to the degeneration of the retinal ganglion cells. To identify the genetic defect in a family with putative ADOA, we performed capture next generation sequencing (CNGS) to screen known retinal disease genes. However, six exons failed to be sequenced by CNGS in optic atrophy 1 gene (OPA1). Sequencing of those exons identified a 4 bp deletion mutation (c.2983-1_2985del) in OPA1. Furthermore, we sequenced the transcripts of OPA1 from the patient skin fibroblasts and found there is six-nucleotide deletion (c.2984-c.2989, AGAAAG). Quantitative-PCR and Western blotting showed that OPA1 mRNA and its protein expression have no obvious difference between patient skin fibroblast and control. The analysis of protein structure by molecular modeling suggests that the mutation may change the structure of OPA1 by formation of an alpha helix protruding into an existing pocket. Taken together, we identified an OPA1 mutation in a family with ADOA by filling the missing CNGS data. We also showed that this mutation affects the structural intactness of OPA1. It provides molecular insights for clinical genetic diagnosis and treatment of optic atrophy.
Similar content being viewed by others

Introduction
Autosomal dominant optic atrophy (ADOA) is the most frequent form of hereditary optic neuropathy. It is characterized by optic nerve pallor, central or centrocecal visual field loss, color vision deficits and progressive loss of visual acuity in the early decades of life due to the degeneration of the retinal ganglion cells1. In most cases, ADOA patients experience a slowly progressive and insidious decrease of their quality of vision, which is irreversible2. The disease prevalence is between 1:12 000 to 1:50 000 in different populations3,4.
Since this disease has no effective treatment, accurate molecular diagnosis is a crucial step for preimplantation genetic diagnosis or prenatal screening.
To date, two genes (OPA1 and OPA3) and three loci (OPA4, OPA5 and OPA8) have been associated with autosomal dominant optic atrophy. OPA2, OPA6 and OPA7 mutations are responsible for X-linked or recessive optic atrophy2. However, OPA1 (NM_130837) is the major gene responsible for ADOA, which was first localized on 3q28 in 1996, followed by the discovery of the OPA1 gene sequence5,6. OPA1 is widely expressed, but most abundant in the retina5. It consists of 31 exons with the last exon as non-coding exon and by alternative splicing from exons 4 to 5b, it produces eight mRNA isoforms7. OPA1 encodes a large GTPase related to dynamins, anchored to the mitochondrial cristae inner membrane, controlling the shape of mitochondrial cristae and keeping their junctions tight during apoptosis8.
OPA1 cleavage has been reported9. Under conditions of stress, it is cleaved by OMA1 (overlapping with the m-AAA protease 1 homolog) to generate shorter isoforms, which trigger mitochondrial fragmentation9. Under high oxidative phosphorylation conditions, it is cleaved by YmeL, the human orthologue of the Yme1 subunit of the yeast i-AAA complex, which stimulates mitochondrial inner membrane fusion10. OPA1 also has been reported to be involved in the maintenance of the cristae structure8,11.
The other gene responsible for ADOA is OPA3 (NM_001017989), which encodes a mitochondrial inner membrane protein with unknown function12. The mutation in OPA3 displayed optic atrophy plus a syndrome consisting of early onset bilateral optic atrophy, later onset spasticity, extrapyramidal signs and cognitive deficit12,13.
In the present study, a patient diagnosed with putative ADOA was recruited. We sought to identify the genetic defect and dissect the molecular consequence of the identified mutation. By capture next generation sequencing (CNGS) and Sanger sequencing, the mutation associated with ADOA in this patient was identified. To dissect the molecular consequence of the mutation, the structure of mitochondria was examined in fibroblasts from patient skin biopsy; the gene and protein expression of OPA1 was tested by quantitative (Q)-PCR and Western blotting, respectively; and the structural changes in OPA1 protein were assessed by molecular modeling.
Results
Clinical data
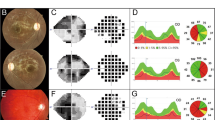
The family in this study comprised six affected individuals from a three generation pedigree (Fig. 1). The proband was a 35 year-old female (marked with a black arrow in Fig. 1). The present vision of the proband is about 20/400. Fundus examination indicated the optic disk in both eyes was well-defined and presented with a bilateral and symmetrical pallor (Fig. 2 A, B, C). 3-D optical coherence tomography (OCT) scans of the optic nerve head revealed the thickness of retinal nerve fiber layer (RNFL) above and below the optic disk and partial temporal sides was obviously attenuated in both eyes (Fig. 2 D, E). OCT scans showed that the macular central fovea had no obvious abnormality in either eye, but RNFL at the (rostral) nasal side and (macular) parafovea region had a tendency to be thinner (Fig. 2 F,G). Flash visual evoked potential (F-VEP) showed P2 wave latencies in both eyes were normal, but amplitude was decreased, indicating defective conduction of the optic nerves (Fig. 2H).

Autosomal dominant optic atrophy (ADOA) pedigree.
Squares and circles symbolize males and females, respectively. Black squares or circles denote affected status, but white squares or circles denote unaffected status. The arrow beside the circles represents the proband.

Clinical data for the left eye.
(A) Optical coherence tomography (OCT) of the optic disc. Significant thickness map of retinal nerve fiber layer (RNFL) indicated in A (thinning parts are showed in red). (B) Eye fundus photos for optic disc. (C), Eye fundus photos. (D–E) The pie chart of RNFL thickness in the left eye. Red parts indicate the significant RNFL thinning. (F) 3D scan section figure of the macular. (G), RNFL thickness map of the macular. RNFL thickness of central fovea is normal, while parafovea and nasal perifovea are significantly thinner. (K) Flash visual evoked potential (F-VEP). N1, N2, P1, P2 indicate the different waves and decreased P2 peak indicates the blocking of optic nerve.
Taken together, based on the clinical manifestations and pedigree, the diagnosis of the patient from this family is putative ADOA.
Identification of the disease-causing gene
To identify the ADOA-causing gene in this family, we performed capture next generation sequencing (CNGS) to screen a panel of genes previously reported to be retinal disease genes (Fig. 3A, Table S1,S4), including the two known genes (OPA1 and OPA3) linked with ADOA. However, we did not detect any disease-causing gene mutations but identified 6 SNPs (Table S2), which includes two novel DNA variants.

Identification of OPA1 mutation.
Mean coverage of Capture Next Generation Sequencing. (A) Mean coverage of 80× over the targeted region was achieved; A portion of the sequenced region is shown. Additional Capture Next Generation Sequencing information is in Table S1, S4. (B) Sequencing results of OPA1 mutation in the patient DNA. This revealed that the patient has a heterozygous mutation. Cloning of the PCR products carrying the mutation and resequenced, confirmed that the mutant allele has 4-base pair deletion.
We rationalize that the mutation may be located in the region of missing coverage or beyond of the panel of the captured genes (Table S1). To rule out the first possibility, we retrieved the data from the CNGS and observed there was no missing coverage in OPA3 but missing coverage of six exons and a low depth (<5) of coverage of ten exons in OPA1(NM_130837). It indicates that no reliable data of more than half of the coding exons (16 of 30) has been obtained from CNGS. To fill in the missing or verify these data, we designed specific primers (Table S3) to amplify these fragments and then sequenced with Sanger sequencing. The mutation analysis led to identification of 4-bp deletion mutation (gAGA, g is part of intron, AGA is part of exon 30, c.2983-1_2985del) in OPA1 (Fig. 3B). We searched for mutations identified to date in the literature and found this mutation has been previously reported in the Italian population14, although the same mutation was named as c.2819-1_2821del due to using another isoform for the mutation annotation. The identification of the same variant in the affected members of the Italian and Chinese pedigrees provides support for its pathogenic nature, since it arose independently in different genetic and ethnic backgrounds.
We also scanned c.2983-1_2985del in two public DNA databases (1000 Genomes Project and ESP6500 Genomes Project) and one in-house exome database containing 1402 samples, but c.2983-1_2985del was absent in all of three DNA databases.
Skin fibroblasts from the ADOA subject have abnormal mitochondrion morphology
Although clinical examination indicated degeneration of the retinal ganglion cells of the ADOA patient, it is difficult to directly study patient-specific ganglion cells in vitro or in vivo. As skin fibroblasts are readily accessible by biopsy, we selected this cell type for further study. The mitochondria of the skin fibroblasts were labeled by mitotracker and then their morphology was observed using confocal microscopy.
As shown in Fig. 4A–D, the morphology of mitochondria in patient-derived fibroblasts and control is dramatically different. Specially, control fibroblasts have spotty distribution of mitochondria (here we define “spotty” as length of mitochondria < 6.9 μm, Fig. 4A, C), however, patient-derived fibroblasts have tubular distribution of mitochondria (“tubular” as length > 6.9 μm, Fig. 4B, D). Statistically, compared with spotty mitochondria in control, only 4.8% ± 2.5% of them from the patient are spotty and the rest (95.2% ± 2.5%) are tubular (Fig. 4A–E). Furthermore, the mitochondrial lengths in the fibroblasts of patient were much larger than that of control (patient, 28.4 ± 5.8 μm versus control, 6.9 ± 1.4 μm, Fig. 4A–D,F). As OPA1 contributes to mitochondrial structure and biogenesis, it would not be surprising if a mutation in this gene alters mitochondrion morphology and therefore contribute to the pathogenesis of ADOA. Interestingly, the growth rate of the patient fibroblasts was much slower than that of control (data not shown), but no obvious skin disorder was observed in this patient.

Mitochondria morphology in skin fibroblasts.
The mitochondrion was labeled using the MitoTracker probe. MitoTracker (red) staining shows aberrant mitochondrial morphology and distribution in fibroblasts with the mutation. (A) Normal fibroblasts (Control) have spotty distribution of mitochondria. (B) Patient-derived fibroblasts have tubular distribution of mitochondria. Nuclei are labeled with DAPI. Scale bars = 20 μm in A, B. (C–D) The higher magnification view of A and B. (E) The different mitochondrial distribution in control and fibroblast of patient, respectively. (F) The average length of mitochondria in fibroblasts from the patient and control. HF-M represents the patient skin fibroblasts (HF-M, human skin fibroblasts with mutation).
Effect of the mutation on OPA1 transcription, protein expression and protein three-dimensional structure
To assess the specific consequence of the c.2983-1_2985del mutation, we extracted mRNA from the patient skin fibroblasts and amplified OPA1 from the cDNA derived from these cells. These amplified fragments were inserted into a cloning vector followed by sequencing the individual clones carrying the amplified fragments. The sequencing results showed the mutant allele was transcribed and the six nucleotide deletion (c.2984-c.2989, AGAAAG) was present (Fig. 5A), which produced a two lysine residue deletion (995–996 deletion) and a missense mutation (Val997Ile) (Fig. 5B). This showed that the genetic deletion mutation in OPA1 creates a new splicing acceptor (Fig. S1). Since both the wild-type and mutant alleles appear to be transcribed, we counted the numbers of wild type and mutant clones we obtained and found no significant difference in their numbers (30 clones of wild type versus 22 mutant) (Fig. 5C). This indicates that there is no significant difference at steady-state transcript levels of OPA1 between the wild type and mutant allele. We then compared the level of OPA1 mRNA expression in the patient skin fibroblasts and control. A slight decrease but no significant of OPA1 mRNA expression was observed in patient skin fibroblasts (Fig. 5D). Western blotting analysis of protein expression extracted from the patient and control fibroblasts showed the same level of OPA1 protein expression (Fig. 5E. F). Furthermore, several different isoforms of OPA1 (approximately 73 KDa, 55 KDa and 37 KDa) were detected.

OPA1 mRNA and protein expression in patient skin fibroblasts.
(A) Sequencing of clones from OPA1 cDNA PCR products. (B) OPA1 mutation analysis. A two residue deletion (995Lys-996Lys) and a missense mutation (Val997Ile) were observed. (C) Relative OPA1 mRNA expression levels in patient skin fibroblasts (HF-M, human skin fibroblasts with mutation) and control. (D) The transcription of the wild type and mutant allele in patient skin fibroblasts. RT-PCR products, harbors the mutation sequence, were cloned into pJET vector; individual clones were picked and then Sanger sequenced. (E) Western blotting for OPA1. (F) The quantitative analysis of Western blotting result was performed using Image J software.
To dissect the molecular mechanism at the level of protein structure, we firstly mapped the mutant residue to the OPA1 protein and found it was located at the C-terminus (Fig. 6A). Multiple sequence alignments were performed and we found that codons 995–997, where the mutation occurred, were located within an extremely conserved region in various species (Fig. 6B).

Mutant OPA1 protein analysis.
(A) Multiple-sequence alignment of OPA1 from different species. The red square indicates the location of the deletion and missense mutation we identified in OPA1 (B) Alpha helical structure of OPA1. The upper sequence is the control; the lower is the OPA1 with deletion/missense mutation. The arrow indicates the location of the deletion/missense mutation in OPA1. The region containing the deletion/missense mutation is predicted to form an alpha helical structure, which was absent from the wild type. (C)In secondary structure predication, the C-ter part (991–1010) forms an alpha helix. The model generated by I-TASSER suggests that this conserved helix (shown in orange), where the mutation is located, protrudes into a pocket. Interruption between this helix and the rest of the pocket will potentially affect the structural intact of the protein.
We predicted the secondary structure of human wild-type and mutant OPA1 using I-TASSER online software. In secondary structure prediction, the C-terminal part of the OPA1 protein (991–1010) forms an alpha helix (Fig. 6B). The 3D model generated by I-TASSER suggests that this alpha helix protrudes into a pocket (Fig. 6C), which will potentially affect the structure of the full protein with functional consequences.
Discussion
Optic atrophy is a group of highly heterogeneous inherited diseases15. Clinical variability, both within and among families with the same disease, often makes recognition and classification difficult. Traditionally, classification has relied on the recognition of similar characteristics and similar patterns of transmission, but genetic analysis now permits the diagnosis of this disease in the absence of family history or in the setting of unusual clinical presentations16. Thus, we initially performed CNGS to identify the genetic defects in this family, but failed to discover any mutation in retinal disease gene. According to the previous study, some captured region may be missing in these tested genes17. Thus, to identify the causative mutation for the disease, filling the missing data may be necessary. Here we showed a case of filling missing CNGS data to search the causative mutation for ADOA, which raises more concern for the clinical application of the next generation sequencing.
In most cases, ADOA presents as a non-syndromic, solely bilateral optic neuropathy, but some patients have associated extra-ocular features including sensory-neural hearing loss, myopathy and peripheral neuropathy18,19,20. At least five mutations in OPA1, p.Arg445His, p.Gly401Asp and p.Leu243*, p.Arg437Glu and p.Ala357Leufs*4 have been reported in patients affected with both optic atrophy and hearing loss21,22,23. However, the proband bearing the same mutation (c.2983-1_2985del) as identified in the present study in Italian family presented optic atrophy plus phenotype, including the mild bilateral temporal pallor of the optic disc, a thinning of the peripapillar bundle nervous layers in both superior and inferior temporal quadrant bilaterally. The Italian patient also displayed a mild bilateral ptosis, ophthalmoparesis, pes cavus, the left side with a decreased/absent Achilles tendon reflex bilaterally. In our study, the patient only had a progressive decrease of vision with no other symptoms, which indicates the same OPA1 mutation may manifest itself differently and be patient dependent and confirms the heterogeneity of this disease. However, further studies are needed to provide insights into the detailed molecular pathogenesis of the retinal ganglion cells affected by this mutation.
The mutation reported in this study occurs at an intron/exon boundary and we would predict it to affect splicing. One possible splice form (S1, Fig. S1–2) is a result of the destruction of the canonical splice acceptor and creating a new splice acceptor insert site. The second possible splice form (S2, Fig. S2) results from skipping exon 30, in other words, a new transcript with a splicing of exon 29 with exon 31 will be generated (Fig. S2). Sequencing of OPA1 cDNA obtained from skin fibroblasts showed that a new splice acceptor site is created resulting in the deletion of two lysine residues and a missense mutation (Fig. S1–2). The same splicing form has been detected in skeletal muscle of the Italian patient14, which reveals the present mutation does change the splicing both in ectoderm and mesoderm derived tissues.
Furthermore, the mutation does not affect the mRNA stability or protein expression. So far, two theories address the cause of ADOA: haploinsufficiency and dominant negative effects24,25,26,27. A dominant negative effect is likely to be the case in this family because both the wild-type and mutant allele appear to have similar levels of steady-state transcription, the patient and control fibroblasts have the same level of OPA1 protein expression.
In summary, this study identified a recurrent mutation in OPA1 in an ADOA family. Our study demonstrates a case of filling missing data of CNGS to search the causative mutation for ADOA, which raises more concern for the clinical application of the next generation sequencing. We also showed the potential molecular consequence of the mutation by protein structural modeling and provided more evidence to support a dominant negative effect to explain the disease pathogenesis. Further detailed functional studies are needed to provide further insights to this inherited disease.
Methods
Patient Recruitment
This study conformed to the tenets of the Declaration of Helsinki. It was approved by the Ethics Committee of Eye Hospital, Wenzhou Medical University. Written informed consent was obtained from the recruited individuals. All experiments were performed in accordance with the approved guidelines. Optical coherence tomography (OCT), flash visual evoked potential (F-VEP) and fundus examination were performed as routine retinal ophthalmic examination. A five ml venous blood sample was drawn into an ethylenediamine tetraacetic acid (EDTA) sample tube. Genomic DNA was extracted from peripheral blood leukocytes using the standard phenol/chloroform extraction protocols. The patient skin fibroblasts and control skin fibroblasts were obtained from skin biopsies.1
Capture Next Generation Sequencing and Bioinformatics Analysis
Capture Next Generation Sequencing was performed as previously described17. The enriched libraries were sequenced on an Illumina Solexa HiSeq 2000 sequencer for paired-end reads of 100 bp. Briefly, using the Solexa QA the cutadapt (http://code.google.com/p/cutadapt/), SOAP aligner, BWA and GATK programs to retrieve and align to identify SNPs and insertions or deletions (InDels). SNPs and InDels were annotated using the exome-assistant program (http://122.228.158.106/exomeassistant). Nonsynonymous variants were evaluated by three algorithms, SIFT (http://sift.jcvi.org/), PolyPhen (http://genetics.bwh.harvard.edu/pph2/) and PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp), to determine pathogenicity. Multiple sequence alignments were performed using ESPript3.0 (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).
Cell culture, mitchondria labeling and cDNA synthesis
The patient skin fibroblasts and control skin fibroblasts were grown in high glucose DMEM with 10% fetal calf serum at 37°C and 5% CO2. The fibroblasts were seeded on coverslips inside a Petri dish filled with medium on day 2, then changed into medium containing 200 nM MitoTrackerR probe (Invitrogen, cat No.M7512) on day 3 and incubated for a further 30 minutes. Cells were then stained with DAPI and observed using a laser Confocal microscope (Zeiss LSM710). The mitochondrial lengths in the fibroblasts were measured using Image J28.
Total RNA were isolated from patient skin fibroblasts and control with TRIzol Reagent (Invitrogen, cat No.15596-026) following the manufacturer's instructions. cDNA synthesis was carried out with M-MLV reverse transcriptase (Invitrogen, cat No. 28025-013). The forward primer 5′-AAGGTGTCAGACAAAGAGAAAATTGAC-3′ and the reverse primer 5′ -TTATTTCTCCTGATGAAGAGCTTCAATG-3′ were used to amplify the coding sequence of OPA1 gene, which harbors the mutation sequence, from the patient fibroblasts, then the PCR fragments were cloned into pJET vector (Fermentas cat No.K1231)and individual clones were picked and then Sanger sequenced.
Quantitative PCR
PCR primers were designed with online software (http://www.idtdna.com/primerquest/Home/Index). For the endogenous control, primers were designed specific to ACTB mRNA (NM28424.1). Sequences of primers used in this study are shown in Table S3. For quantitative PCR, the cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 60°C for 60 s. qPCR data was captured and analyzed using StepOne software (ver. 2.0). qPCR performed using SYBR® Premix Ex Taq™ II (TaKaRa, cat NO RR820A). The OPA1 relative mRNA expression level in the normal and patient fibroblasts were obtained by normalization to ACTB using the formula 2ˆ-(CTgene minus CTcontrol). All qPCR data was performed in triplicate.
Western blotting
For Western blotting, total proteins were extracted from the patient and control skin fibroblasts using RIPA buffer, then fractionated in a 10% SDS-PAGE gel and transferred to PVDF membranes. OPA1 was detected using a mouse anti-OPA1 monoclonal antibody (Santa Cruz, cat No.SC323936) followed by anti-mouse IgG, coupled to IRDye 800CW (Licor Biosciences, cat No. 926-32210). GAPDH was detected using a rabbit anti-GAPDH monoclonal antibody (Bioworld, cat No. AP0063), followed by anti-rabbit IgG, coupled to IRDye 680 (Licor Biosciences, cat No. 926-68071). Western blotting data was analyzed using Image J software28.
References
Johnston, P., Gaster, R., Smith, V. & Tripathi, R. A clinicopathologic study of autosomal dominant optic atrophy. Am J Ophthalmol 88(5), 75–868 (1979).
Lenaers, G. et al. Dominant optic atrophy. Orphanet J Rare Dis 7, 1–12 (2012).
Kivlin, J. D., Lovrien, E. W., Bishop, D. T. & Maumenee, I. H. Linkage analysis in dominant optic atrophy. Am J Hum Genet 35, 1190–5 (1983).
Kjer, B., Eiberg, H., Kjer, P. & Rosenberg, T. Dominant optic atrophy mapped to chromosome 3q region. II. Clinical and epidemiological aspects. Acta Ophthalmol Scand 74, 3–7 (1996).
Alexander, C. et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26, 211–215 (2000).
Delettre, C. et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26, 207–210 (2000).
Delettre, C. et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 109, 584–591 (2001).
Frezza, C. et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126, 177–189 (2006).
Anand, R. et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204, 919–929 (2014).
Mishra, P., Carelli, V., Manfredi, G. & Chan, D. C. Proteolytic cleavage of opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab 19, 630–641 (2014).
Olichon, A. et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278, 7743–7746 (2003).
Reynier, P. et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet 41, e110 (2004).
Grau, T. et al. A novel heterozygous OPA3 mutation located in the mitochondrial target sequence results in altered steady-state levels and fragmented mitochondrial network. J Med Genet 50, 848–858 (2013).
Ranieri, M. et al. Optic atrophy plus phenotype due to mutations in the OPA1 gene: two more Italian families. J Neurol Sci 315, 146–149 (2012).
Reynier P Fau - Amati-Bonneau, P. et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet 41, e110 (2004).
Biousse1, N. J. N. V. Hereditary optic neuropathies. Eye 18, 1144–1160 (2004).
Zhang, X. et al. Molecular diagnosis of putative stargardt disease by capture next generation sequencing. PLoS One 9, e95528 10.1371/journal.pone.0095528. eCollection 2014 (2014).
Huang, T., Santarelli, R. & Starr, A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res 1300, 97–104 (2009).
Yu-Wai-Man, P., Trenell, M. I., Hollingsworth, K. G., Griffiths, P. G. & Chinnery, P. F. OPA1 mutations impair mitochondrial function in both pure and complicated dominant optic atrophy. Brain 134, e164–e164 (2011).
Yu-Wai-Man, P. et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–86 (2010).
Ferre, M. et al. Molecular screening of 980 cases of suspected hereditary optic neuropathy with a report on 77 novel OPA1 mutations. Hum Mutat 30, E692–705 (2009).
Shimizu, S. et al. A novel mutation in the OPA1 gene in a Japanese patient with optic atrophy. Am J Ophthalmol 135, 256–257 (2003).
Amati-Bonneau, P. et al. The association of autosomal dominant optic atrophy and moderate deafness may be due to the R445H mutation in the OPA1 gene. Am J Ophthalmol 136, 1170–1171 (2003).
Marchbank, N. J. et al. Deletion of the OPA1 gene in a dominant optic atrophy family: evidence that haploinsufficiency is the cause of disease. J Med Genet 39, e47 (2002).
Amati-Bonneau, P. et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131, 338–351 (2008).
Hudson, G. et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131, 329–337 (2008).
Olichon, A. et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol 211, 423–430 (2007).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We thank the families for their participation in this project. We are indebted to Dr. Jinyu Wu (Institute of Genomic Medicine, Wenzhou Medical University, China) for scanning the present mutation in in-house exome database and Dr.Ji Sun (Department of Pharmacology, University of Washington, Seattle, WA, USA) for structural modeling. This work was supported by grants from the Chinese National Program on Key Basic Research Project (973 Program, 2013CB967502, FG), Natural Science Foundation of China (81201181/H1818, FG), Zhejiang provincial & Ministry of Health research fund for medical sciences (WKJ2013-2-023, FG), Medical Scientific Projects from Health Bureau of Zhejiang Province (2011ZDA016, WL), Wenzhou Medical University Grant (QTJ 12011, FG and KYQD131102, LPZ).
Author information
Authors and Affiliations
Contributions
F.G. conceived the idea and supervised the research. L.P.Z., W.S., L.M.S., X.Z., L.L.C., X.L.G., Y.F.W., W.L., W.Z. and Z.B.J. collected the samples and performed the experiments. Q.J.M., J.Q. and F.G. performed data analyses, F.G. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary File
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Zhang, L., Shi, W., Song, L. et al. A recurrent deletion mutation in OPA1 causes autosomal dominant optic atrophy in a Chinese family. Sci Rep 4, 6936 (2014). https://doi.org/10.1038/srep06936
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06936
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
