
Abstract
We examine improvements to the linear mixed model (LMM) that better correct for population structure and family relatedness in genome-wide association studies (GWAS). LMMs rely on the estimation of a genetic similarity matrix (GSM), which encodes the pairwise similarity between every two individuals in a cohort. These similarities are estimated from single nucleotide polymorphisms (SNPs) or other genetic variants. Traditionally, all available SNPs are used to estimate the GSM. In empirical studies across a wide range of synthetic and real data, we find that modifications to this approach improve GWAS performance as measured by type I error control and power. Specifically, when only population structure is present, a GSM constructed from SNPs that well predict the phenotype in combination with principal components as covariates controls type I error and yields more power than the traditional LMM. In any setting, with or without population structure or family relatedness, a GSM consisting of a mixture of two component GSMs, one constructed from all SNPs and another constructed from SNPs that well predict the phenotype again controls type I error and yields more power than the traditional LMM. Software implementing these improvements and the experimental comparisons are available at http://microsoft.com/science.
Similar content being viewed by others


Introduction
There has been a great deal of interest in statistical methods for genome-wide association studies (GWAS). While linear or logistic regression have been commonly used for this task, the need to move beyond these models has become clear. One important motivation for more sophisticated models is the existence of confounding structure, including population structure and family relatedness. Recently, the linear mixed model (LMM) has emerged as the model of choice to correct for such confounding structure1,2,3,4,5,6,7,8,9. Despite its rapid acceptance, however, there remain concerns about its use and several improvements have been proposed.
One suggested improvement is the inclusion of principal components (PCs) as covariates to better capture population structure8. Another proposed improvement is to use only a subset of single nucleotide polymorphisms (SNPs) for inclusion in the LMM4,5,6,7,10. In particular, the LMM relies on an estimate of the genetic similarity matrix (GSM), which encodes the pairwise similarity between every two individuals in the data set. These similarities are estimated from SNPs or other genetic variants. While traditionally all available SNPs are used to estimate the GSM, researchers have considered using a subset, chosen in at least two different ways.
In one approach, SNPs are chosen such that they are roughly equally spaced across the genome4. The idea behind this approach is that linkage disequilibrium (LD) among the SNPs mitigates the need to use all of them. One motivation underlying this approach is computational efficiency. Namely, when the number of selected SNPs is less than the sample size of the data, then the computation of P values becomes linear in sample size, rather than quadratic4. We shall refer this to form of subsetting as LD sampling.
A second approach to subsetting is based on a mathematical equivalence between the LMM and linear regression4,7,11. Specifically, an LMM is equivalent to a form of linear regression in which the SNPs that determine the GSM in the LMM view are covariates in the linear-regression view. The linear-regression view suggests including in the GSM only those covariates that are correlated to the phenotype5,7. The inclusion of causal or tagging SNPs could improve GWAS power by reducing the model misspecification that would otherwise result from their exclusion. Inclusion of SNPs that tag confounding structure could help correct for confounding by effectively using these SNPs as covariates5,10. Perhaps most importantly, omitting irrelevant SNPs, which introduce noise, could amplify these benefits7. We refer to this form of subsetting as SNP selection. A second motivation for SNP selection is the potential for computational efficiency as just mentioned for LD sampling. We note that, despite these motivations, there has been debate about its usefulness12.
Here, we thoroughly investigate these potential improvements under conditions spanning a wide range of population structure and family structure. In our experiments, we also consider how the distribution of effect sizes affects the usefulness of these improvements. Although we offer some theoretical insights, this work is primarily empirical. For these empirical investigations, we employ three types of data sets: synthetic SNPs with synthetic phenotypes, real SNPs with synthetic phenotypes and real SNPs with real phenotypes.
We concentrate our investigations on real-valued phenotypes and data that are randomly ascertained. While an LMM can often be successfully applied to binary phenotypes (e.g., containing cases and controls)3,13,14, in preliminary studies not presented here, we find that our results generally do not extend to situations with substantial ascertainment bias.
In our experiments, we first generate a suite of data sets having various degrees of confounding structure and distributions of effect sizes. Next, we apply each of the models under investigation as described in Table 1 to each of the data sets, performing GWAS (computing SNP-phenotype association P values) for each combination of model and data set. Finally, we evaluate empirical type I error rate and power for each model based on the P values obtained over the suite of datasets.
Table 1 summarizes the main results. Selection on its own did not live up to its promise. In the presence of population structure, family relatedness, or both, selected SNPs did not sufficiently correct for confounding in that the resulting model failed to control for type I error. Interestingly, however, when combined with a second fixed or random effect, we found that the use of SNP selection controls for type I error and increases power. Specifically, when only population structure was present, using selection in combination with PCs included as (fixed-effect) covariates, yielded more power than the traditional LMM. When family structure was present (with or without population structure), SNP selection was again useful, but only when used with a new improvement—namely, a mixture of two GSMs, one constructed from all SNPs and another constructed from SNPs identified by selection. This GSM-mixture model both controlled type I error well and yielded more power than the traditional LMM. This model also performed well when no confounding structure was present and when only population structure was present.
Another notable finding was that the improvements to power afforded by SNP selection manifested most strongly when the number of causal SNPs was low and they had large effect sizes. Finally, on data with real SNPs (where LD was present), we found that replacing a GSM based on all SNPs with one based on LD sampling improved run time without degradation in type I error or power.
Results
We evaluated various GWAS models across three broad sets of experiments: those involving synthetic SNPs and synthetic phenotypes, those involving real SNPs and synthetic phenotypes and those involving real SNPs and real phenotypes. As noted in the introduction, we focused on data that were randomly ascertained. Also, we concentrated on genome-wide association analyses that test for associations between a single SNP and a phenotype, although the methods we considered should also be applicable to a variety of association tests, such as those between sets of SNPs and a phenotype15. Herein, we use the term GWAS to refer to univariate association analyses only.
Synthetic SNPs and phenotypes
We generated synthetic SNPs and phenotypes under four settings: no population structure or family relatedness, population structure only, family relatedness only and both population structure and family relatedness. We generated each data set with M = 50,000 SNPs and N = 4,000 individuals, typical of many GWASs. (Note that ref. 12 suggests that there are effectively 60,000 independent SNPs in the human genome.) We generated SNPs such that there was no physical linkage to avoid any confusion about the identity of true causal SNPs. In our data generation, we varied the degree of population structure, family structure, number of causal SNPs and signal strength over a wide range of plausible parameters, including ones yielding strong confounding from population structure and family relatedness, so as to challenge and thereby uncover weaknesses of the various models examined. Results varied depending on the presence of population structure and on the presence of family relatedness, so we consider the four possible cases separately.
No population or family relatedness
For each data set, we generated SNPs with a minor allele frequency (MAF) sampled uniformly from the range [0.05, 0.5]. For each individual, a continuous phenotype was then constructed by generating causal and noise components and summing them. The causal component was generated from a linear model with a varying number of causal SNPs C. The causal SNPs were normalized to have mean zero and variance one and the effect sizes were drawn identically and independently from a Gaussian distribution with mean zero and variance  . The independent noise component was generated independently and identically from a Gaussian distribution with mean zero and variance
. The independent noise component was generated independently and identically from a Gaussian distribution with mean zero and variance  .
.
Parameter values used in our simulations were as follows:
-
Number of causal SNPs: 10, 50, 100, 500, 1000
-
Narrow-sense heritability (causal signal)
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 Three data sets for each possible combination of parameters were generated, yielding 3 × 5 × 6 = 90 data sets. Note that different random seeds were used to generate each set of SNPs so that no two sets were the same.
We used these datasets to measure empirical type I error rate and power for three models: linear regression (Linreg), an LMM with a GSM based on all SNPs (LMM(all)) and an LMM with a GSM based on SNP selection (LMM(select)). See Methods for a detailed description of the LMM model. As motivated in the introduction by the correspondence between an LMM and regression, SNPs were selected so as to maximize prediction accuracy on the phenotype. In particular, SNPs were identified by searching over multiple sets of SNPs to identify those that maximized out-of-sample prediction accuracy as measured by the log likelihood of the phenotype under the LMM. To keep the search practical, we ordered SNPs for each fold by their increasing univariate linear-regression P values and then considered increasing numbers of SNPs in this order (see Methods for details). This approach is known as marginal regression16. The computational complexity of the algorithm is O(N2M).
For each of the three models, we measured empirical type I error rate (the proportion of non-causal SNPs deemed significant) as a function of P-value threshold. In addition, we measured empirical power (the proportion of causal SNPs deemed significant) as a function of empirical type I error (Figure 1; note that a fourth method shown in the figure, LMM(all + select), will be introduced in the following section). Results are shown for different numbers of causal SNPs. All models controlled type I error well. Furthermore, LMM(select) yielded the most power, especially when the number of causal SNPs was small (and thus the effect sizes were large). That LMM(select) had more power than Linreg is not surprising when thinking about the LMM as linear regression with selected SNPs as covariates. Namely, conditioning on selected SNPs reduces noise in the phenotype. That LMM(select) had more power than LMM(all) when there were few causal SNPs is also expected, as presumably the use of all SNPs in the GSM obfuscated the true causal signal, a phenomenon called “dilution”7.

Empirical type I error rate and power for no population or family relatedness with purely synthetic data.
Type I error rate is plotted as a function of P value cutoff α. Each point represents the average type I error rate or power across 18 data sets with different degrees of signal (narrow-sense heritability). Shading on the curves for type I error rate represent the 95% confidence interval assuming type I error is controlled. All power curves that are visibly separated have significant differences between them. For example, comparing power for Linreg and LMM(all) for 10 causal SNPs at a type I error of 10−3, the P value from a two-sided binomial test applied to the number of true positives is 0.03.
One interesting finding was that SNP selection would select all SNPs in many data sets when only a relatively small number of the SNPs in the generating data were causal (Figure 2). One explanation is that, as the number of causal SNPs increases for a fixed narrow-sense heritability, the signal in each SNP decreases. Therefore, even for a relatively small number of causal SNPs (e.g., less than 1000), the SNP selection algorithms may not be able to detect the signal at the individual-SNP level, thus finding all SNPs to be optimal. (See ref. 17 for a theoretical discussion.) Indeed, when we used 1000 causal SNPs and increased narrow-sense heritability beyond 0.4, less than all SNPs (in fact, less than 1000) were selected.

Box plots showing number of SNPs selected and mixing weight as a function of the number of causal SNPs with purely synthetic data.
The first column shows log10 of the number of SNPs selected by LMM(select). The highest point corresponds to the selection of all SNPs. The second and third columns show the number of selected SNPs and mixing weights for LMM(all + select). A mixing weight of 1 corresponds to using a GSM based only on SNP selection. A mixing weight of 0 corresponds to using a GSM based only on all SNPs.
Population structure but no family relatedness
To introduce population structure, all SNPs were generated from the Balding-Nichols model18 with a 50:50 population ratio, a baseline MAF sampled uniformly from [0.05, 0.5] and a value for Wright's FST that varied across the generated data sets. The data was otherwise generated as described for the previous setting of no population structure.
Parameter values used in these simulations were as follows:
-
Number of causal SNPs: 10, 50, 100, 500, 1000
-
Narrow-sense heritability
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 -
Degree of population structure FST: 0.005, 0.01, 0.05 and 0.1
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 Three data sets for each possible combination of parameters were generated, yielding 3 × 5 × 6 × 4 = 360 data sets. As under the previous setting of no population structure and no family relatedness, different random seeds were used to generate each set of SNPs so that no two sets were the same.
For each of the three models considered previously (Linreg, LMM(all), LMM(select)), we examined exclusion and inclusion of PCs as fixed-effect covariates. In Supplemental Material, we compared two methods for estimating PCs. In one approach, PC estimation was guided by the accuracy of phenotype prediction, similar to the approach of refs. 19,20,21 for estimating PCs for linear and logistic regression. In the second approach, PC estimation was guided by the prediction accuracy of PCs on SNPs rather than the phenotype, based on a Probabilistic Principal Components (PPC) model22. This second approach yielded better control of type I error and power and herein we concentrate on only this approach. The algorithm, described in Methods, has a computational complexity O(N2M) in the simple case where all N PCs are computed.
The inclusion of PCs had differing effects on the performance of the models (Supplementary Figure 1). LMM(all) controlled type I error well, whether or not PCs were included as fixed effects and inclusion did not affect power. In contrast, for Linreg, inclusion of PCs led to control of type I error, consistent with the results in ref. 23 and had little effect on power. Furthermore, the inclusion of PCs led to control of type I error and improved power for LMM(select), as was recently reported in an independent investigation24.
One way to understand these results is through consideration of the graphical-model structure25 for our data-generation process (Figure 3a). Here, l is a hidden (latent) variable denoting which of the two Balding-Nichols populations the individual is in, y is the phenotype and c and s with subscripts denote the causal and non-causal SNPs, respectively. The graphical-model structure conveys assertions of conditional independence among the variables represented. For example, when l is unobserved, there are open paths between the SNP variables, reflecting induced correlation among the SNPs. In contrast, if l were observed, these paths would be closed, reflecting the lack of correlation among SNPs within each population. Ref. 25 describes generally how to determine correlations from the graph based on what is and is not observed.

Graphical models for the data-generation process.
The variable l is hidden (latent) and corresponds to confounding structure, either population structure of family relatedness. The variables c and s with subscripts correspond to causal and non-causal SNPs, respectively.
The graph offers a simple explanation for the inflation of P values (i.e., the lack of type I control) when using Linreg. In particular, because l is unobserved, there are open paths in the graph from the non-causal SNPs to y, indicating a correlation between the non-causal SNPs and y. The graph also offers two possible explanations for why LMM(all) without PCs leads to good control of type I error. One is that, as described in the introduction, the use of LMM(all) is equivalent to a form of linear regression where all SNPs condition the phenotype. By conditioning on all the SNPs and thus the causal ones, we block all paths from l to y, again making the non-causal SNPs and y independent. The other is that a GSM based on all SNPs may accurately represent the latent variable l, again blocking the paths from non-causal SNPs to y. Later in this section, we will see evidence that at least the second explanation holds true. The graph also explains results for LMM(select). Namely, the SNP selection algorithm presumably failed to select some of the causal SNPs, resulting in paths from the non-causal SNPs to y. To validate this hypothesis, we estimated a GSM from only the true causal SNPs (those generating the phenotype), observing no inflation (Supplemental Figure 2). In addition, when PCs were included with LMM(select), they accurately represented the latent variable (see Supplemental Material), thus blocking the paths from non-causal SNPs to y and yielding control of type I error.
Similar to what we observed in the case of no population or family relatedness, SNP selection selected fewer than all SNPs in most data sets only when there were a few causal SNPs with large effect size (Figure 2). Furthermore, in these situations, LMM(select) with PCs yielded more power than LMM(all) (Supplemental Figure 2).
So far, our simulations have assumed that confounding has resulted only from differences in SNP allele frequencies between populations. In practice, however, there may be additional confounding effects due to correlations between the Balding-Nichols populations (represented by l in Figure 3) and the phenotype. For example, environmental effects may differ between the two populations. To investigate such additional confounding effects, we simulated data as before, but now added a direct correlation between l and y as shown in Figure 3b (see Methods). To do so, we first generated 100 additional SNPs in the same way as the other SNPs except with a fixed FST of 0.2. We then used these 100 SNPs to generate a component added to the phenotype with variance  , such that
, such that  . These SNPs were used only in the generation of the phenotype. They were excluded from the GWAS analysis.
. These SNPs were used only in the generation of the phenotype. They were excluded from the GWAS analysis.
Consistent with results just presented, we found that the use of PCs led to good control of type I error. In addition, we found that use of a GSM based on all SNPs led to good control (Supplemental Figure 3). This observation indicates that a GSM based on all SNPs can accurately represent (i.e., block paths through) the hidden variable l.
In summary, on purely synthetic data with population structure but no family relatedness, we found that LMM(select) yielded better GWAS performance than LMM(all), but only when PCs were used as covariates.
Family relatedness but no population structure
We generated data as described for the first setting of no confounding structure, except that we created family relatedness by mating randomly selected synthetic individuals, producing 10 offspring per parent pair. The fraction of offspring in the population was varied across the generated data sets so as to yield a degree of inflation for Linreg similar to that in the population-structure setting. In a single mating, the genotype of the child was constructed by selecting one copy of the genotype from the mother and one copy from the father. Matings were performed in two passes, creating equal numbers of offspring in each pass. In the first pass, each mother and father pair was selected from the same population. In the second pass, each pair was selected randomly from the existing set of individuals, possibly from different populations.
Parameter values used in these simulations were as follows:
-
Number of causal SNPs: 10, 50, 100, 500, 1000
-
Narrow-sense heritability
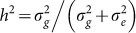 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 -
Degree of family relatedness; fraction of individuals belonging to a family: 0.5, 0.6, 0.7, 0.8, 0.9
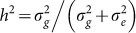 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 Three data sets for each possible combination of parameters were generated, yielding 3 × 5 × 6 × 5 = 450 data sets. Again, different random seeds were used to generate each set of SNPs so that no two sets were the same.
Under this setting, we evaluated Linreg + PCs, LMM(all) and LMM(select) + PCs. Linreg + PCs and LMM(select) + PCs failed to control type I error, in contrast to the setting with population structure, whereas LMM(all) controlled type I error (Figure 4 and Supplementary Figure 4). Again, we can understand these results in terms of the graphical-model structure for the data-generation process, which is that of Figure 3a where now the hidden variable corresponds to family relatedness. In terms of this graph, inflation observed for Linreg + PCs (also seen in Ref. 8) indicates that the use of PCs as fixed effects failed to block the paths through l from non-causal SNPs to y. Similarly, inflation observed for LMM(select) + PCs indicates that neither PCs as fixed effects nor selected SNPs blocked the paths through l. Only LMM(all) blocked all paths from the non-causal SNPs to y, either by conditioning on all SNPs, having a GSM that fully captures family relatedness l, or both. Later in this section, we will see evidence supporting at least the second explanation.

Empirical type I error rate and power with and without population structure (PS) and family relatedness (FR) with purely synthetic data.
Type I error rate is plotted as a function of P value cutoff α. Each point represents the average type I error rate or power across multiple data sets with varying numbers of causal SNPs and varying degrees of heritability, population structure and family relatedness.
Turning to power, we found that LMM(select) performed best (Supplementary Figure 4). Thus, interestingly, no model performed best in terms of both type I control and power. Based on this observation, we developed a new LMM model having a GSM made up of a mixture of two GSMs ((1 − π) K0 + π K1), one based on all SNPs (K0) and one based on SNP selection (K1). Methods provides a detailed description of the algorithm for creating this model, called LMM(all + select).
Because we included a mixture component based on all SNPs, the algorithm considered only a relatively small number of SNPs for the selected component. Nonetheless, because one of the components was based on all SNPs, the computational complexity of this algorithm was the same as that of LMM(select): O(N2M). Note that this model is closely related to those in refs. 26,27 and is also related to the model of ref. 10 who added SNPs as fixed effects identified with forward-backward selection conditioned on a GSM estimated from all SNPs.
This new model yielded the benefits of both a GSM based on selected SNPs and one based on all SNPs. It controlled type I error and had power equal to that of an LMM with selected SNPs (Figure 4 and Supplementary Figure 4). As before, the power advantage was greater when a relatively small number of the SNPs in the generated data were causal. The model also performed well in the settings of no confounding structure and population structure only (Figures 1 and 4 and Supplemental Figure 2).
Interestingly, the number of SNPs selected first increased and then decreased with the number of causal SNPs (Figure 2). Presumably, the decrease was due to the fact that one of the components of the mixture GSM is based on all SNPs and that this component can well represent larger numbers of causal SNPs with smaller effect size, as we have seen under the previous settings of confounding structure. This explanation is consistent with the mixing weights π estimated for the mixture GSM (Figure 2). Namely, when there were a small number of causal SNPs with large effect size, the estimated mixing weight was high, favouring the GSM based on SNP selection. When there were a large number of causal SNPs with small effect sizes, the estimated mixing weight was low, favoring the GSM based on all SNPs.
As before, we explored additional confounding due to family relatedness, corresponding to a direct arc from l to y in Figure 3b. Such confounding could result from, for example, family-related environmental effects. To create this additional confounding structure, we first generated 100 additional SNPs subject to the same family relatedness as the other SNPs and then used these 100 SNPs to generate a component added to the phenotype with variance  , such that
, such that  .
.
Linreg and LMM(select) failed to control type I error due to the open paths from non-causal SNPs to y. In contrast, LMM(all) and LMM(all + select) controlled type I error (Supplementary Figure 5), indicating that a GSM based on all SNPs is capable of blocking the paths through l—that is, a GSM based on all SNPs is capable of capturing family relatedness.
Population structure and family relatedness
We generated data as described for the setting of population structure and no family relatedness, except we added family relatedness by mating randomly selected individuals as described in the previous section. Parameter values used in these simulations were as follows:
-
Number of causal SNPs: 10, 50, 100, 500, 1000
-
Narrow-sense heritability
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 -
Degree of population structure; FST: 0.005, 0.01, 0.05 and 0.1
-
Degree of family relatedness; fraction of individuals belonging to a family: 0.5, 0.6, 0.7, 0.8, 0.9
 : 0.1, 0.2, 0.3, 0.4, 0.5, 0.6
: 0.1, 0.2, 0.3, 0.4, 0.5, 0.6 Three data sets for each possible combination of parameters was generated, yielding 3 × 5 × 6 × 4 × 5 = 1800 data sets. Again, no two sets of SNPs were the same.
Using this data, we examined four models: Linreg + PCs, LMM(all), LMM(select) + PCs and LMM(all + select). The model LMM(all + select), which performed best for the setting of family relatedness without population structure, also performed best here (Figure 4 and Supplementary Figure 6). These results indicate that the inclusion of all SNPs as part of the mixture GSM led to good control of type I error for both forms of confounding structure, consistent with our findings for family relatedness alone and population structure alone. Furthermore, the inclusion of selected SNPs as part of the mixture GSM led to improved power, again most notably so when there were a small number of causal SNPs with large effect size (Supplementary Figure 6). Finally, as we saw for the setting of family structure alone, the number of SNPs selected first increased and then decreased with the number of causal SNPs (Figure 2).
Real SNPs and synthetic phenotypes
We next compared models using data sets based on real SNPs and synthetic phenotypes. We used real SNPs to determine whether our results on synthetic SNPs would hold under more realistic conditions, including those where SNPs were in LD. We continued to use synthetic phenotypes to maintain a gold standard as to which SNPs were causal.
We used real SNPs from three cohorts, two from human studies—the Northern Finnish Birth Cohort from 1966 (Finnish) and CIDR Visceral Adiposity Study (VAS)—and one from a mouse cross (Mouse) (see Methods). These data contain various degrees of population structure and family relatedness. From a hierarchical clustering performed on each of these three data sets (Figure 5), we see that Finnish contains little population structure or family relatedness, VAS contains mostly population structure as illustrated by the broad bands of similarity and the mouse data contains both forms of confounding structure as illustrated by the combination of broad and narrow bands. We generated the phenotype in essentially the same manner as for purely synthetic data sets, always using h2 = 0.5.

The GSM for three real SNP data sets.
Each point in the matrix corresponds to the similarity between a pair of individuals. Lighter colors correspond to greater similarity. The ordering was obtained by a hierarchical clustering, as indicated by the dendrograms on the axes, where different colors reflect substantially different clusters.
To understand our strategy for measuring empirical type I error rate and power, it is useful to first understand an effect that can lead to loss in power. When computing an association P value for a given SNP, that SNP and SNPs nearby from the GSM should not be included4,5,14. If these SNPs are included, then (as follows from the linear-regression view of the LMM) they effectively become covariates in the test and reduce power. This effect is called proximal contamination4,5. In the experiments with synthetic SNPs, which contained no LD due to recombination, it was sufficient to exclude only the single SNP tested from the GSM in order to avoid proximal contamination. We did so using the method in ref. 5. In the experiments with real SNPs in LD, we could have used a similar approach and excluded SNPs from the GSM that were close to the SNP being tested. However, this approach was somewhat computationally expensive. Thus, instead we excluded from the GSM all SNPs on the chromosomes being tested, as has been described previously4,5,12. Specifically, we measured type I error rate with SNPs from chromosome 1 and power with SNPs from chromosome 2, after first sampling causal SNPs from all but chromosome 1 and constructing the GSM with SNPs from all but chromosomes 1 and 2 (repeating this procedure multiple times to obtain good estimates). One potential problem with this approach is that leaving out chromosome 2 from the GSM when measuring type I error rate on chromosome 1 would lead to open paths from the non-causal SNPs on chromosome 1 to the phenotype (through causal SNPs on chromosome 2). These exclusions could then result in inflated P values due not to a poor model, but rather to this expedient approach of avoiding proximal contamination. We will examine this potential problem in more detail after considering the main results of the analysis, but for purposes of evaluating the various models, we avoided this problem by making sure that no causal SNPs were on chromosome 2 when evaluating type I error rate on chromosome 1.
We applied the models Linreg, Linreg + PCs, LMM(select), LMM(select) + PCs, LMM(all) and LMM(all + select) to each of these data sets, yielding results that were consistent with our findings on the purely synthetic data. In particular, for the Finnish SNPs, which had little population structure or family relatedness, all models controlled type I error and models using SNP selection had more power than models that did not (Figure 6 and Supplementary Figure 7). For the VAS SNPs, which contained mostly population structure, all models except Linreg and LMM(select) controlled type I error and again models using SNP selection had more power than models that did not (Figure 6 and Supplementary Figure 8). For the Mouse SNPs, which exhibited both forms of confounding structure, only LMM(all) and LMM(all + select) controlled type I error and LMM(all + select) had the most power, presumably because it was the only model that both used SNP selection and controlled type I error (Figure 6 and Supplementary Figure 9).

Empirical type I error rate and power for three real SNP data sets and synthetic phenotypes with 10 causal SNPs.
Each point represents the average type I error rate or power across multiple synthetic phenotypes (400 for Finnish and AVS and 4,000 for Mouse). In the Finnish power plot, methods that include select have greater power than those that do not.
Returning to the potential problem of leaving out one chromosome to avoid proximal contamination, we investigated this problem by repeating our experiments on Mouse data but now allowing causal SNPs to be sampled from chromosome 2 when evaluating type I error rate on chromosome 1. The result was that the leave-out-one-chromosome approach did indeed lead to inflated P values (Supplementary Figure 10). Consequently, although we were able to avoid the bad effects of leaving out one chromosome in our synthetic experiments, we recommend that, in practice, the method only be used when avoiding proximal contamination with a small window around the tested SNP is not computationally feasible.
Finally, LD among the SNPs in this data allowed us to investigate the usefulness of replacing a GSM estimated from all SNPs with one estimated after LD sampling, as first suggested in ref. 4. We did so for the Mouse SNPs, where we had found a GSM based on all SNPs to be most needed for control of type I error. A sample of only one fourth of the available 10,000 SNPs yielded good control of type I error (Figure 7), suggesting that, at least for this SNP data, LD sampling can be an effective approach to improving the run time of GWAS.

Empirical type I error rate and power for phenotypes synthetically generated from SNPs from the Mouse data with 10 causal SNPs.
GSMs were estimated from SNPs sampled uniformly across the genome (every kth SNP). Each point represents average type I error rate or power across 4,000 synthetic phenotypes.
Real SNPs and phenotypes
We applied our models to fully real data to check for consistency with our findings on synthetic data. In general, evaluation of real data is difficult, because the gold standard (i.e., the identity of the causal SNPs) is unknown. Nonetheless, some real data sets have a bronze standard—a validated collection of causal SNPs or SNPs that tag causal ones—making a partially informative analysis possible, with the limitation that the list of validated SNPs is incomplete. In this work, we analysed the Finnish data set with phenotypes low density lipoprotein (LDL), high density lipoprotein (HDL) and triglycerides (Trig) and the VAS data set with phenotype BMI. The causal SNPs and associated loci for the bronze standard were obtained from the NHGRI GWAS catalog (http://www.genome.gov/gwastudies). We examined four models: Linreg, LMM(select) + PCs, LMM(all) and LMM(all + select).
To get a sense of type I error rate, we counted apparent false positive loci. A locus was considered a false positive if it contained a below threshold SNP more than two million bases away from any catalog SNP. To get a sense of power, we counted the number of loci deemed to be true positives. A locus was considered a true positive if it was in the catalog and contained a below threshold SNP. There were no significant differences (Supplementary Table 1). For the VAS data set, LMM(select) + PCs selected all SNPs. This observation is consistent with our results from synthetic data, as the BMI phenotype is thought to have many causal SNPs. For the Finnish data set, all methods yielded zero false positives at thresholds of 5 × 10−7 and 5 × 10−8, consistent with our understanding that the Finnish data has little population structure or family relatedness.
Discussion
Traditionally, when an LMM is used for GWAS, its GSM is estimated from all available SNPs. In this work, we have evaluated potential improvements to this approach on a broad set of data, both synthetic and real.
One potential improvement, building a GSM based on selected SNPs that well predict the phenotype failed rather dramatically. In particular, when population structure, family relatedness, or both were present, this approach failed to control for type I error. Presumably, SNPs sufficient for good prediction are not sufficient for good GWAS performance. These results are in contrast to our previous findings7, which used less realistic simulations. Nonetheless, when SNP selection was used in combination with other improvements, it proved useful. Specifically, in the presence of population structure alone, SNP selection in combination with PCs used as covariates controlled type I error and also yielded more power than the traditional approach. In all settings, with or without population structure or family relatedness, a mixture of two GSMs, one constructed from all SNPs and another constructed from SNPs identified by SNP selection both controlled type I error and yielded more power than the traditional LMM. Furthermore, the improvements to power afforded by SNP selection were the strongest when some SNPs had a large effect size.
Of course, when analyzing real data, there will be uncertainty about the distribution of effect sizes and about how much population structure and family relatedness are present. Consequently, we recommend using the mixture GSM when feasible. One drawback of this approach is computational expense: run time is O(N2M), where N and M are the sample size and number of tested SNPs, respectively. One observation that could mitigate this problem is that the GSM component based on all available SNPs can be replaced with one based on a set of SNPs sampled across the genome (LD sampling). Depending on the degree of LD among the SNPs, a subject for further investigation, the sample could in principle be small enough such that the number of sampled SNPs k would be less than N, yielding a O(NMk) computational complexity4. A second benefit implied by the potential effectiveness of LD sampling is that the measurement of a large number of SNPs (e.g., whole-genome sequencing) would be unnecessary for building the GSM.
Another way to improve run time would be to remove closely related individuals (if any) from the analysis and then employ LMM(select) + PCs. This approach has several drawbacks. First, there would be a loss in power due to the removal of individuals. Also, inflation could remain due to distant family relatedness (cryptic relatedness). In addition, the approach may not work well in a setting where population structure deviates from the idealized Balding-Nichols structure employed in our investigations. Finally, although its run time is less than GSM-mixture approach, its computation complexity is no better: O(N2M).
We also found that avoiding proximal contamination by excluding SNPs from the GSM that are in the same chromosome as the SNP being tested can lead to inflated P values. When feasible, we recommend excluding SNPs in a small window around the SNP being tested. Ref. 5 provides a relatively efficient algorithm for doing so.
Interestingly, we found that a GSM based on all SNPs (or LD-sampled SNPs) could account for population structure just as well as PCs. Consequently, if SNP selection picks all SNPs, then there is no need to add PCs to the LMM. We note that ref. 8 showed that adding PCs could be beneficial, but only in rare situations where some SNPs are unusually differentiated.
Our experiments suggest that good GWAS performance across multiple settings requires two model components: a component based on all (or LD sampled) SNPs to control type I error and a component based on selected SNPs to improve power. These two components are random effects in our mixture GSM, but ref. 10 has had success with a model wherein one component is a GSM based on all SNPs and the other component is a set of fixed-effect SNPs identified by forward-backward selection. (The use of a fixed-effect component rather than a random-effect component should be particularly useful when the number of SNPs with large effect size is very small.) Other approaches for creating two-component models including alternative methods for selecting SNPs (e.g., refs. 28,29,30), seem worthy of investigation.
Finally, we emphasize that our empirical investigations covered only randomly ascertained data. Initial investigations suggest that many of our conclusions do not apply when there is strong ascertainment bias as can be found in case-control studies12. Identifying improvements for the ascertained case is a subject for further investigation.
Software implementing the traditional LMM and the improvements discussed here as well as the simulations and evaluations are available at http://microsoft.com/science.
Methods
Description of real data
The Finnish data comes from the NFBC1966 Study31 and was pre-processed as described in ref. 32, yielding 328,517 SNPs for 5,256 individuals. No covariates were used, as they were regressed out during pre-processing. The VAS data (dbGap phs000169.v1.p1) was filtered with individual missingness < 0.1, MAF > 0.05, SNP missingness < 0.1 and Hardy-Weinberg disequilibrium P values > 0.00001, yielding 720,036 SNPs for 2,802 individuals. The real BMI phenotype was analyzed with age, gender, race, site, smoking amount, drinking amount and education as covariates. The Mouse data and its pre-processing is described in ref. 33 yielding 10,150 SNPs for 1,940 individuals.
The linear mixed model
An LMM decomposes the variance associated with phenotype y into the sum of a linear additive genetic and residual component. The distribution of y is given by

where X is the N × Q matrix of Ν individuals and Q covariates (e.g., gender, age) including an offset term, β is the Q × 1 vector of fixed effects, I is the N × N identity matrix, K is the GSM of size N × N (determined from a set of SNPs),  is the variance of the genetic component and
is the variance of the genetic component and  is the variance residual component.
is the variance residual component.
As discussed in the main text, the LMM model (equation 1) is equivalent to a form of linear regression. In particular, consider the phenotype as a linear regression of SNPs Z (N × S) on phenotype, with mutually independent effect sizes α distributed  . Assuming the values for each SNP are standardized, the log likelihood can be written as
. Assuming the values for each SNP are standardized, the log likelihood can be written as

Identifying  , we recover equation (1). Note that, when
, we recover equation (1). Note that, when  , K is called the realized relationship matrix (RRM)34. The RRM is commonly used in genetic studies11 and we do so here. For a detailed discussion of the LMM, see ref. 35.
, K is called the realized relationship matrix (RRM)34. The RRM is commonly used in genetic studies11 and we do so here. For a detailed discussion of the LMM, see ref. 35.
As discussed, we concentrated on genome-wide association analyses that test for associations between a single SNP and a phenotype. When using the LMM to compute a P value for the association between a test SNP and the phenotype, we used an F-test where parameters were estimated using restricted maximum likelihood (REML). The value for  estimated for the null model was also applied to the alternative models3,36. We assumed an additive effect of a SNP on the phenotype. In particular, the value of a SNP for a given individual was encoded as the number of minor alleles of the SNP for that individual (0, 1 or 2).
estimated for the null model was also applied to the alternative models3,36. We assumed an additive effect of a SNP on the phenotype. In particular, the value of a SNP for a given individual was encoded as the number of minor alleles of the SNP for that individual (0, 1 or 2).
Description of algorithms
The algorithm for SNP selection was as follows:
-
1
Create random train-test partitions of the data samples (corresponding to individuals).
-
2
For each partition
-
a
Use the training data to compute univariate linear-regression P values on each SNP.
-
b
Order the SNPs by increasing P value.
-
c
For numSNPs in {0, 1, 2, 4, …, 1024, all} (the default values), use the first numSNPs as features for the LMM:
-
i
Optimize the parameters of the LMM using REML.
-
ii
Use the LMM to compute the predictive log likelihood of the test data (the log joint probability density of the test data given the training data).
-
i
-
a
-
3
Choose the value of numSNPs that maximizes the sum over the partitions of the predictive log likelihood of the test data.
In step 1, we use a 90%–10% train-test partition of the data with enough partitions such that there are 10,000 samples in the test sets overall. In practice, this number of samples leads to the selection of a similar number of SNPs for different random seeds. This default applies to the other method that uses random train-test partitions as well. The most time consuming step in the algorithm is the evaluation of the predictive log likelihood when all SNPs are used in step 2c. The computational complexity of this step is O(N2M). In the experiments with synthetic SNPs and phenotypes, we used the search grid {0, 1, 5, 10, 20, 50, 100, 500, 1000, 2000, 5000, 10000, all} in step 2c. In the experiments with real SNPs and synthetic phenotypes, we used the search grid {0, 1, 3, 10, 32, 101, 322, 1024, all}. In the experiments with real data, we used the default values above.
The algorithm for estimating PCs was a follows:
-
1
Remove individuals that are closely related. Our default removes individuals until no two individuals have an estimated kinship coefficient from a GSM computed from all genome-wide variants of less than 0.1.
-
2
One dimension of the matrix of SNPs indexes multivariate samples, while the other indexes variables of the samples. To avoid problems due to the high dimensionality of the SNPs of each individual, treat the SNPs as samples and the individuals as variables.
-
3
Partition the samples into subsamples for cross-validation. Partition SNPs across chromosomes to reduce correlation between the partitions. When evaluating prediction accuracy for a given subsample, the subsample is used for testing, while the other subsamples are used for training. Each train-and-test constitutes a fold.
-
4
For numPCs = 0
-
a
For each fold
-
i
Use the training data to compute maximum likelihood estimates for a PPC model.
-
ii
Compute the log likelihood of the test data according to this model.
-
i
-
a
-
5
Repeat step 4 with increasing numPCs until either (1) the sum over the folds of log likelihood decreases twice in a row or (2) this sum first increases and the decreases below the starting value.
-
6
Select the PPC model that maximizes the sum over the folds of the log likelihood.
-
7
Project the individuals who were removed in step 1 onto the subspace defined by the optimal PPCA model (see Supplementary Material).
The algorithm for creating LMM(all + select) was as follows:
-
1
Create random train-test partitions of the individuals.
-
2
For each partition
-
a
Use the training data to compute P values on each SNP based on an LMM with a GSM using all SNPs.
-
b
Order the SNPs by increasing P value.
-
c
For numSNPs in {0, 1, 2, 4, …, 128} (the default values), use the first numSNPs as features for the LMM:
-
i
Optimize the parameters of the LMM including the mixing weight π by REML.
-
ii
Use the LMM to compute the predictive log likelihood of the test data (the log probability density of the test data given the training data).
-
i
-
a
-
3
Choose the value of numSNPs that maximizes the sum over the partitions of the predictive log likelihood of the test data.
Because we included a mixture component based on all SNPs, the algorithm considered only a relatively small number of SNPs for the select component (see step 2c).
References
Yu, J. et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–8 (2006).
Kang, H. M. et al. Efficient control of population structure in model organism association mapping. Genetics 178, 1709–23 (2008).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–54 (2010).
Lippert, C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–5 (2011).
Listgarten, J. et al. Improved linear mixed models for genome-wide association studies. Nat. Methods 9, 525–6 (2012).
Listgarten, J., Lippert, C. & Heckerman, D. FaST-LMM-Select for addressing confounding from spatial structure and rare variants. Nat. Genet. 45, 470–1 (2013).
Lippert, C. et al. The benefits of selecting phenotype-specific variants for applications of mixed models in genomics. Sci. Rep. 3, 1815; 10.1038/srep01815 (2013).
Price, A., Zaitlen, N., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–63 (2010).
Zhou, X. & Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 44, 821–4 (2012).
Segura, V. et al. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–30 (2012).
Hayes, B. J., Visscher, P. M. & Goddard, M. E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. (Camb). 91, 47–60 (2009).
Yang, J., Zaitlen, N. A., Goddard, M. E., Visscher, P. M. & Price, A. L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 46, 100–6 (2014).
Agresti, A. Categorical Data Analysis. (Wiley, 2002).
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Listgarten, J. et al. A powerful and efficient set test for genetic markers that handles confounders. Bioinformatics 29, 1526–33 (2013).
Genovese, C. & Wasserman, L. A Comparison of the Lasso and Marginal Regression. J. Mach. Learn. Res. 13, 2107–2143 (2011).
Helmbold, D. & Long, P. On the Necessity of Irrelevant Variables. J. Mach. Learn. Res. 13, 2145–2170 (2012).
Balding, D. J. & Nichols, R. A. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96, 3–12 (1995).
Novembre, J. & Stephens, M. Interpreting principal component analyses of spatial population genetic variation. Nat. Genet. 40, 646–9 (2008).
Lee, S., Wright, F. A. & F. Z. Control of population stratification by correlation-selected principal components. Biometrics 67, 967–974 (2011).
Hoffman, G. E. Correcting for population structure and kinship using the linear mixed model: theory and extensions. PLoS One 8, e75707 (2013).
Tipping, M. E. & Bishop, C. M. Probabilistic Principal Component Analysis. Analysis 1–13 (1999).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–9 (2006).
Tucker, G., Price, A. L. & Berger, B. Improving the Power of GWAS and Avoiding Confounding from Population Stratification with PC-Select. Genetics 197, 1045–1049 (2014).
Pearl, J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. (Morgan Kaufmann, 1988).
Zhou, X., Carbonetto, P. & Stephens, M. Polygenic Modeling with Bayesian Sparse Linear Mixed Models. PLoS Genet. 9 (2013).
Speed, D. & Balding, D. J. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res. (2014).
Hoggart, C. J., Whittaker, J. C., De Iorio, M. & Balding, D. J. Simultaneous analysis of all SNPs in genome-wide and re-sequencing association studies. PLoS Genet. 4, e1000130 (2008).
Rakitsch, B., Lippert, C., Stegle, O. & Borgwardt, K. A Lasso multi-marker mixed model for association mapping with population structure correction. Bioinformatics 29, 206–214 (2013).
Dolejsi, E., Bodenstorfer, B. & Frommlet, F. Analyzing genome-wide association studies with an FDR controlling modification of the Bayesian Information Criterion. PLoS One 9, e103322 (2014).
Sabatti, C. et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat. Genet. 41, 35–46 (2009).
Fusi, N., Lippert, C., Lawrence, N. D. & Stegle, O. Genetic Analysis of Transformed Phenotypes. arXiv (2014).
Valdar, W. et al. Genetic and environmental effects on complex traits in mice. Genetics 174, 959–84 (2006).
Hayes, B. J., Visscher, P. M. & Goddard, M. E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. (Camb). 91, 47–60 (2009).
Lippert, C. Linear mixed models for genome-wide association studies. Ph.D. Diss. Tuebingen (2013).
Zhang, Z. et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42, 355–60 (2010).
Acknowledgements
We thank Alkes Price and Noah Zaitlen for useful discussions. The NFBC1966 Study is conducted and supported by the National Heart, Lung and Blood Institute (NHLBI) in collaboration with the Broad Institute, UCLA, University of Oulu and the National Institute for Health and Welfare in Finland. This manuscript was not prepared in collaboration with investigators of the NFBC1966 Study and does not necessarily reflect the opinions or views of the NFBC1966 Study Investigators, Broad Institute, UCLA, University of Oulu, National Institute for Health and Welfare in Finland, or the NHLBI. Funding support for the CIDR Visceral Adiposity Study was provided through the Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, NIA. The CIDR Visceral Adiposity Study includes a genome-wide association study funded as part of the Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, NIA. Assistance with phenotype harmonization and genotype cleaning as well as with general study coordination was provided by Heath ABC Study Investigators.
Author information
Authors and Affiliations
Contributions
C.W. designed research, conducted experiments, contributed analytic tools, analyzed data and wrote the paper. C.L. designed research, conducted experiments, contributed analytic tools, analyzed data and wrote the paper. O.W. designed research, conducted experiments, analyzed data and wrote the paper. N.F. designed research and wrote the paper. C.K. and R.D. contributed analytic tools. J.L. designed research, contributed analytic tools and wrote the paper. D.H. designed research, conducted experiments, contributed analytic tools, analyzed data and wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
C.W., C.L., N.F., C.K., R.D., J.L. and D.H. were employed by Microsoft while performing this work.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Widmer, C., Lippert, C., Weissbrod, O. et al. Further Improvements to Linear Mixed Models for Genome-Wide Association Studies. Sci Rep 4, 6874 (2014). https://doi.org/10.1038/srep06874
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06874
This article is cited by
-
Diallel panel reveals a significant impact of low-frequency genetic variants on gene expression variation in yeast
Molecular Systems Biology (2024)
-
Characterization of novel loci controlling seed oil content in Brassica napus by marker metabolite-based multi-omics analysis
Genome Biology (2023)
-
Genomic prediction with whole-genome sequence data in intensely selected pig lines
Genetics Selection Evolution (2022)
-
Rare and population-specific functional variation across pig lines
Genetics Selection Evolution (2022)
-
Genome-wide association study and its applications in the non-model crop Sesamum indicum
BMC Plant Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
