
Abstract
To realize cardiac regeneration using human induced pluripotent stem cells (hiPSCs), strategies for cell preparation, tissue engineering and transplantation must be explored. Here we report a new protocol for the simultaneous induction of cardiomyocytes (CMs) and vascular cells [endothelial cells (ECs)/vascular mural cells (MCs)] and generate entirely hiPSC-engineered cardiovascular cell sheets, which showed advantageous therapeutic effects in infarcted hearts. The protocol adds to a previous differentiation protocol of CMs by using stage-specific supplementation of vascular endothelial cell growth factor for the additional induction of vascular cells. Using this cell sheet technology, we successfully generated physically integrated cardiac tissue sheets (hiPSC-CTSs). HiPSC-CTS transplantation to rat infarcted hearts significantly improved cardiac function. In addition to neovascularization, we confirmed that engrafted human cells mainly consisted of CMs in >40% of transplanted rats four weeks after transplantation. Thus, our HiPSC-CTSs show promise for cardiac regenerative therapy.
Similar content being viewed by others


Introduction
Cardiovascular disease remains the leading cause of death in the Western world1,2. Despite significant advances in therapeutic modalities, such as heart transplantation or ventricular assist device implantation and risk-reduction strategies, a substantial disease burden remains3. This health problem has prompted research into new therapeutic strategies including regenerative medicine with stem cells4,5,6. Among various stem cell populations, pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), possess outstanding capacity for cardiac regeneration due to their potential of infinite expansion and efficient differentiation into most somatic cell lineages7,8. Nevertheless, many obstacles, such as poor engraftment of the injected cells to the heart, have inhibited the clinical translation of cardiac cell therapies based on these stem cell populations9,10.
We have developed a cell-sheet system using a culture surface grafted with a temperature-responsive polymer, poly (N-isopropylacrylamide) (PIPAAm), which enables cell sheet collection without enzymatic digestion and allows us to easily generate a transplantable tissue-like structure11,12,13. Previously, we reported a transplantation study in rat infarcted hearts using cardiac tissue sheets bioengineered with mouse ESC-derived defined cardiac cell populations with cardiomyocytes (CMs), endothelial cells (ECs) and mural cells (MCs; vascular smooth muscle cells and pericytes)11. All of these populations were systematically induced from ESC-derived Flk1 (also designated as vascular endothelial cell growth factor [VEGF] receptor-2)-positive mesoderm cells as common cardiovascular progenitors14,15,16. In that previous study, we showed clear functional recovery through paracrine effects, such as neovascularization, that were mainly mediated by donor CM-derived angiogenic factors such as VEGF. VEGF secretion from donor CMs was highly enhanced by the co-existence of ECs, indicating the importance of cellular interactions between CMs and non-myocytes in cell sheet functions.
Here we extend our cardiac cell sheet strategy towards a more clinical direction using human iPSC-derived cell sheets. We hypothesized that cardiac tissue sheets, including cardiovascular cell populations induced from human iPSCs (hiPSC-CTSs), could show high potential for ameliorating the cardiac dysfunction that follows myocardial infarction (MI).
Results
Simultaneous induction of CMs and vascular cells from human iPSCs
Human iPSCs were simultaneously differentiated toward CMs and vascular cells (ECs and MCs) with a modified directed differentiation protocol (Fig. 1a,b). This modification is based on our previous report, which described a monolayer culture-based efficient CM differentiation protocol17. In that protocol, the gene expression level of cardiac mesoderm and/or progenitor genes (KDR/ISL1) peaks on differentiation day 5 (d5) and the addition of Dkk1 (a canonical Wnt antagonist) during d5-7 enhanced CM differentiation from mesoderm cells (Fig. 1a, left). This time, we attempted vascular cell induction together with CMs using an angiogenic cytokine, VEGF, which we have reported induces EC differentiation from mouse ESC-derived Flk1-positive mesoderm cells14. The addition of VEGF instead of Dkk1 during d5-15 resulted in the simultaneous induction of ECs along with CMs, which was not observed in our previous method (Fig. 1 and Supplementary Fig. 1). The cellular component of the cardiovascular cell populations on d15 was 76.1 ± 16.9% for cTnT (cardiac troponin-T)-positive CMs, 10.6 ± 4.8% for vascular endothelial (VE)-cadherin (CD144)-positive ECs and 10.9 ± 14.4% for platelet-derived growth factor receptor beta (PDGFRβ; CD140b)-positive MCs according to flow cytometry (n = 13, VEGF 50 ng/ml, Fig. 1c). These results indicate that this stage-specific modification can control the direction of the differentiation from “exclusive CMs” to “CMs plus vascular cells” upon the appropriate proportional induction of each cardiovascular cell population. We also confirmed that during the differentiation protocol, the TRA-1-60-positive undifferentiated human iPSC component was diminished to 1.2 ± 0.8% of total cells on d15 from approximately 80% of cells on d0 (Fig. 1c).

Simultaneous induction of CMs and vascular cells from human iPSCs.
(a) Schematic diagram of cardiovascular cell induction protocols. Defined cardiovascular cell populations (cardiomyocytes [CMs], endothelial cells [ECs] and vascular mural cells [MCs]) are systematically differentiated from human iPSCs. Left panel: a protocol to induce exclusively CMs. Right panel: a modified protocol to induce a variety of cardiovascular cells. (b) Schematic representation of the cardiovascular cell induction protocol. (c) Flow cytometry analysis of cellular components on d15 (n = 13, VEGF 50 ng/ml). hiPSC, human induced pluripotent stem cell; VEGF, vascular endothelial cell growth factor; Dkk1, Dickkopf-related protein 1; MEF-CM, mouse embryonic fibroblast conditioned medium; bFGF, basic fibroblast growth factor; ActA, Activin A; BMP4, bone morphogenetic protein 4; cTnT, cardiac troponin-T; VE-cad, vascular-endothelial cadherin; PDGFRb, platelet-derived growth factor receptor beta.
Generation of hiPSC-Cardiac Tissue Sheets (CTSs)
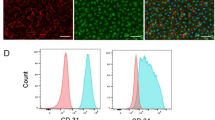
We then generated cell sheets with hiPSC-derived cardiovascular cells. We continued culturing cells until d15 before generating cell sheets (3–6 days after the appearance of beating cells), which brought less contamination of undifferentiated human iPSCs among the cultured cells. We collected the differentiated cells at d15, plated them onto 12-multiwell temperature-responsive culture plates (1.0 × 106 cells/well, UpCell, CellSeed, Tokyo, Japan) and re-cultured them for an additional 4 days. Temperature reduction successfully provided re-assembled self-pulsating cell sheets (hiPSC-CTSs) (Fig. 2a,b and Supplementary Video 1). The CTS consisted of 3-4 cell layers with intact stratified structure of collagen adjacent to the cell components (Fig. 2c). Immunostaining for CMs (cTnT-positive) showed that CMs were dominantly distributed within the sheet (Fig. 2c). The CTS consisted of CMs (72.2 ± 16.5% of total cells), ECs (5.7 ± 5.1%) and MCs (20.2 ± 22.3%) according to flow cytometry (n = 13) (Fig. 2d). Undifferentiated cells were 1.1 ± 0.7% of total cells in the sheet (n = 13) (Fig. 2d). The slight change in cellular components compared to those before plating onto UpCell plates is possibly due to differences in attachment efficiency, cell death and proliferation efficiency among each population. The total cell count of the sheet was 7.44 ± 1.78 × 105 on average (n = 13). These results indicate that a cardiac tissue-like structure, including CMs and vascular components, was successfully generated from hiPSCs only. As a comparison, we also attempted to generate a sheet construct from purified CMs. We performed immunomagnetic selection for VCAM1 (vascular cell adhesion molecule 1; CD106)-positive cells at d15, which are considered pure CMs (>95%)17. We plated the purified CMs onto UpCell plates, however, these cells failed to form sheet structures even with sufficient cell numbers for confluence after 4 days of culture (Supplementary Fig. 2). These results suggest that non-myocytes are essential for generating a cell sheet structure by re-assembling cardiac cell populations.

Generation of hiPSC-cardiac tissue sheets (CTSs).
(a) Schematic diagram of hiPSC-CTS formation. Cardiac cell populations (CMs, ECs and MCs) are differentiated from hiPSCs, collected, then re-cultured to construct tissue sheets. (b) Macroscopic appearance of hiPSC-CTS. Left panel: 1-ply. Right panel: 3-ply. (c,d) Tissue sheet components. (c) Histological analyses. Upper left panel: Hematoxylin-Eosin (HE) staining. Upper right panel: Sirius-red staining (red) for a cross-section showing intact extracellular matrix. Lower panel: Immunostaining of a sheet for cTnT (CMs, red) and DAPI (cell nuclei, blue). (d) Cellular components analyzed by flow cytometry (n = 13). DAPI, 4, 6 diamidino -2-phenylindole. Scale bars: 1 cm in b, 100 μm in c (lower panel) and 20 μm in c (upper panels).
HiPSC-CTS transplantation ameliorates cardiac dysfunction after rat sub-acute myocardial infarction
Three hiPSC-CTSs were layered to form a 3-sheet structure in vitro (Fig. 2b) and transplanted into an athymic nude rat sub-acute MI model11,18. All transplanted and sham-operated rats (n = 19 for each group) survived the 8-week post-transplant observation period with no tumor formation. We followed cardiac function with an echocardiogram during this 8-week period (n = 19) (Fig. 3)19. HiPSC-CTS transplantation dramatically restored anterior wall contraction, which had been reduced after MI induction (Fig. 3a). Transplantation significantly improved parameters for left ventricle (LV) systolic function, fractional shortening (FS) and fractional area change (FAC) (Fig. 3b,c) and also significantly recovered systolic thickening of the infarct wall (Fig. 3d). The percentage of akinetic endocardial length in the whole LV endocardial circumference (AL) was approximately 20% in the transplantation group after MI induction (almost equal with that of the sham operation group), but recovered to become almost undetectable after 8 weeks of transplantation, whereas it gradually increased in the sham operation group (Fig. 3e). Although the LV diastolic diameter increased in both groups, the extent of dilation was significantly smaller in the transplantation group compared to the sham operation group (Fig. 3f). We also examined the extent of fibrosis after transplantation. Sirius red staining revealed that hiPSC-CTS transplantation limited the extent of fibrosis at 4 weeks after transplantation compared to the sham operation group (Supplementary Fig. 3). All of these findings indicate that hiPSC-CTS transplantation improved LV systolic function through the attenuation of LV remodeling after MI, which was also observed in our previous report using mouse ESC-derived CTS transplantation11.

Echocardiograms of cardiac functions after hiPSC-CTS transplantation to rat MI.
(a) Representative M-mode image of sham-operated rat (left) and CTS transplanted rat (Tx; right). Note that the infarct anterior wall started to contract 2–4 weeks after transplantation in Tx rat. Dotted lines: boundary of anterior or posterior wall of LV. (b) Fractional shortening (FS). (c) Fractional area change (FAC). (d) Systolic thickening. (e) Akinetic length (AL). (f) Diastolic diameter of LV (mm). *P<0.05, **P<0.01 and ***P<0.001 (two-way ANOVA and Tukey's test vs. PreTx). †P<0.05 and ‡P<0.001 (two-way ANOVA). n = 19. PreMI, Pre-induction of MI; PreTx, Pre-transplantation; Tx2w, Tx4w, Tx2mo, 2 weeks, 4 weeks and 2 months after transplantation, respectively; LV, left ventricle.
Engraftment of human iPSC-derived cells and neovascularization following hiPSC-CTS transplantation
Next, we traced the extent of cell engraftment after transplantation of hiPSC-CTS using immunostaining for human nuclear antigen (HNA), which is positive for transplanted human cells and negative for rat recipient cells. At an early phase after transplantation (3 days after transplantation; Tx-d3), distinct clusters of engrafted human CMs positive for HNA and cTnT were observed in most examined rats (Fig. 4a). In contrast to our previous results with mouse ESCs, in which almost no engraftment of transplanted cells was observed at a chronic phase (Tx-d28), engrafted human cell clusters that consisted mainly of CMs were still observed in over 40% of transplanted rats even at a chronic phase (4 of 9 rats; 44.4%). The engrafted area reached 24.7 ± 15.7% of the infarcted area (ranged from 5.5% to 44.0%) (Fig. 4b). Von Willebrand factor (vWF) staining for ECs demonstrated a prominent accumulation of vWF-positive ECs in close proximity to the engrafted CMs at Tx-d3 (Fig. 4a), indicating an angiogenic mechanism mediated by the engrafted human iPSC-derived CMs similar to that mediated by mouse ESC-derived CMs11. The angiogenic effect resulted in a significant increase in capillary density within the infarcted area at Tx-d28 (Supplementary Fig. 4). We further investigated vascular formation in the engrafted area at Tx-d28. Even though we could not confirm graft-originated vascular formation in the engrafted area, we found recipient-originated vascular luminal structures (vWF+/HNA−) penetrating the engrafted human cell clusters (Fig. 4c). These results suggest that the functional benefits after hiPSC-CTS transplantation should be mediated by the synergistic enhancement of a fair engraftment of hiPSC-derived CMs supported by blood supply and a neovascularization mediated by transplanted human CM-derived angiogenic paracrine effects.

Engraftment of human iPSC-derived cells and neovascularization following hiPSC-CTS transplantation.
(a) Immunostaining with consecutive sections on Tx-d3. Left panel: double staining for vWF (ECs, green) and cTnT (CMs, red) and DAPI. Right panel: double staining for vWF (ECs, green) and HNA (human cell nuclei, red) and DAPI. (b,c) Immunostaining on Tx-d28. (b) Double staining for cTnT (CMs, green) and HNA (human cell nuclei, red) and DAPI. Upper panels: representative staining for engrafted area. Right panel: higher magnification view of left panel (square). Lower panel: quantification of engrafted area. The ratio of the engrafted area (area in the broken magenta line) and the MI area (area in the broken white line) is shown. (c) Double staining for vWF (ECs, green) and HNA (human cell nuclei, red) and DAPI. Tx-d3 and d28: 3 and 28 days after Tx, respectively. Scale bars: 2 mm in b (lower panel), 500 μm in b (upper left panel), 200 μm in a and 100 μm in b (upper right panel) and c.
Discussion
In the present study, we developed a novel human iPSC differentiation protocol for cardiovascular cells, generated fully hiPSC-engineered CTSs and showed efficient effects and mechanisms for cardiac regeneration after sheet transplantation into a rat MI model. Enhanced neovascularization and sheet engraftment suggests the importance of the co-existence and interaction between CMs and non-myocytes for stem cell-based cardiac regeneration. There exist several reports describing highly human iPSC-derived CM-enriched cell sheet transplantation for a porcine MI model with positive functional outcomes20,21. Although we could not directly compare the therapeutic effects of cell sheets with “exclusive CMs” and those with “CMs plus vascular cells” due to an inability to form pure CM sheets (Supplementary Fig. 2), the addition of vascular cells, that is, CTSs consisting of “CMs plus vascular cells”, is expected to bring better outcomes than those consisting of “exclusive CMs” considering the results presented here and those from the transplantation of mouse ESC-derived CTSs11. Further attempts to control the cell distribution for a more organized tissue structure might enhance sheet functions and beneficial outcomes after transplantation22.
Though this study extends our previous mouse ESC-based study11 to human iPSCs, we found significant differences, including a considerable volume of survived cell clusters 4 weeks after transplantation in the present work that was not observed in the mouse study. Although the mechanisms of the increased engraftment remain unclear, there are a number of possible potent endogenous survival mechanisms that may explain the difference, including anti-apoptosis, promotion of cell survival, cell homing or angiogenesis23. Indeed, moderate vascularization in grafted tissues from the recipient was observed (Fig. 4c), although we could not demonstrate a clear contribution by the donor-derived vascular graft. Bioengineering strategies that pre-vascularize the graft tissue might enhance the vascular connection between the host and graft for better long-term engraftment24.
All transplanted rats survived the 8-week observation period with no accidental death suggesting that there might be no lethal arrhythmic event even on rats with fair engraftment of human CMs. It was reported that human ESC-derived CM transplantation attenuated ventricular tachycardia after myocardial infarction25. This advantageous effect might also be possible with the present human iPSC-derived cell transplantation. Assessment of the proarrhythmic potential of the iPSC-CTS transplantation is critical before this method reaches clinical application. However, we were unable to obtain direct evidence of any electrical integration between the grafted and host tissues or synchronous contractions of the grafted CMs with the host hearts due to the considerably different beating rates between rodent and human hearts. Therefore, larger animal models that have similar intrinsic beating rates with human hearts may be preferred. We also found no tumor formation during the observation period. At the same time, longer observation periods would be desirable to further certify the safety of this iPSC-derived cell transplantation therapy26. We anticipate continued evolution in the technical generation of “safer” iPSCs with minimal risk of tumor formation are required prior to the initiation of human iPSC-based cardiac cell therapy27,28. Although several safety issues still remain to be resolved, our results demonstrate the promise of hiPSC-derived cardiac tissue sheet transplantation in cardiac regeneration after MI.
Methods
All protocols were approved by the Kyoto University Animal Experimentation Committee and performed in accordance with the guidelines for Animal Experiments of Kyoto University, which conforms to Japanese law and the Guide for the Care and Use of Laboratory Animals prepared by the Institute for Laboratory Animal Research, U.S.A. (revised 2011).
Human iPSC culture and differentiation
Human iPSCs [4-factor (Oct3/4, Sox2, Klf4 and c-Myc) line: 201B6 and 6-factor episomal plasmid vector (Oct3/4, Sox2, Klf4, L-Myc, LIN28 and Glis1) line: 836B3] were established previously8,27. 201B6 was used as representative human iPSCs representative in all experiments unless stated otherwise. Human iPSC culture method was previously reported in detail17,29. These cells were adapted and maintained on thin-coat matrigel (growth factor reduced, 1:60 dilution; Invitrogen, Eugene, USA) in mouse embryonic fibroblast conditioned medium (MEF-CM) supplemented with 4–5 ng/mL human basic fibroblast growth factor (hbFGF; WAKO, Osaka, Japan). Cells were passaged as small clusters once every 4–6 days using CTK solution (0.1% collagenase IV, 0.25% Trypsin, 20% knockout serum replacement (KSR) and 1 mM CaCl2 in phosphate buffered saline (PBS)).
Cardiovascular cell differentiation was induced as previously reported17,29 with some modifications, as shown in Fig. 1b. Cells were detached following a 3–5 min incubation with Versene (0.48 mM EDTA solution; Invitrogen) and seeded onto matrigel-coated plates at a density of 100,000 cells/cm2 in MEF-CM with 4 ng/mL bFGF for 2–3 days before induction. Cells were covered with matrigel (1:60 dilution) on the day before induction. To induce cardiac differentiation, we replaced MEF-CM with RPMI+B27 medium (RPMI1640, 2 mM L-glutamine, 1× B27 supplement without insulin) supplemented with 100 ng/mL of Activin A (ActA; R&D, Minneapolis, USA) for 24 hours, followed by 10 ng/mL human bone morphogenetic protein 4 (BMP4; R&D) and 10 ng/mL bFGF for 4 days without culture medium change. The culture medium was subsequently replaced with RPMI+B27 supplemented with VEGF165 (WAKO) and culture medium was refreshed every other day. VEGF165 was used at a concentration of 50 ng/ml in all experiments unless stated otherwise. Beating cells appeared at day 9–12. “n” values in the main text mark the number of times the differentiation experiments with the present protocol were independently performed. For the cardiomyocyte differentiation protocol without VEGF (Supplementary Fig. 1), we used RPMI+B27 supplement with insulin and 100 ng/mL of Dkk1(R&D) on d5-7 and refreshed culture medium every other day with RPMI+B27 supplement with insulin.
Human iPS cell-derived cardiac tissue sheet formation
Cells after differentiation (d15) were dissociated by incubation with Accumax (Innovative Cell Technologies, San Diego, USA) and plated onto a gelatin-coated 12-multiwell UpCell at 2.6 × 105 cells/well with 1 mL of attachment medium (AM; alpha minimum essential medium (αMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 5 × 10−5 M 2-mercaptoethanol) containing VEGF165 and a ROCK inhibitor (Y-27632, 10 μM; WAKO). After two days in culture, the culture medium was replaced with RPMI1640 medium containing 2 mM L-glutamine, 10% FBS and VEGF165. After two more days in culture, cells were moved to room temperature. Within 15–30 minutes, cells detached spontaneously and floated in the media as a monolayer cell sheet.
Flow cytometry, cell sorting and fluorescent microscopy
Cells after hiPSC differentiation and generated cell sheets were dissociated by incubation with Accumax and stained with one or a combination of the following surface markers: anti-VCAM1 conjugated with allophyco-cyanin (APC), clone STA, 1:200 (BioLegend, San Diego, USA); anti-PDGFRβ conjugated with phycoerythrin (PE), clone 28d4, 1:100 (BD, Franklin Lakes, USA); anti-VE-cadherin conjugated with fluorescein isothiocyanate (FITC), clone 55-7h1, 1:100 (BD); and anti-TRA-1-60 conjugated with FITC, clone Tra-1-60, 1:20 (BD). To eliminate dead cells, cells were stained with the LIVE/DEAD fixable Aqua dead cell staining kit (Invitrogen). For cell surface markers, staining was carried out in PBS with 5% FBS. For intracellular proteins, staining was carried out on cells fixed with 4% paraformaldehyde (PFA) in PBS. Cells were stained with the anti-cardiac isoform of Troponin T (cTnT) (clone 13211, Thermo Fisher Scientific, Waltham, USA) labeled with Alexa-488 using Zenon technology (Invitrogen) (1:50). The staining was performed in PBS with 5% FBS and 0.75% Saponin (Sigma, St. Louis, USA). Stained cells were analyzed and sorted on an AriaII flow cytometer (BD). Data were collected from at least 5,000 events. For magnetic activated cell sorting (MACS; Miltenyi, Bergisch Gladbach, Germany), cells were stained with anti-VCAM1 antibody conjugated with APC (BioLegend) followed by anti-APC microbeads (Miltenyi). Data were analyzed with DIVA software (BD). For fluorescent microscopy, hiPSC-CTSs were stained for cTnT (anti-cardiac isoform of cTnT, Thermo Fisher Scientific; 1:500) with DAPI (4, 6 diamidino -2-phenylindole; Invitrogen). Anti-mouse Alexa 546 (Invitrogen) was used as a secondary antibody. Stained sheets were photographed with an all-in-one fluorescent microscopic system, Biorevo BZ-9000 (Keyence, Osaka, Japan).
Animal model preparation, transplantation and echocardiogram
Male athymic nude rats (F344/N Jcl-rnu/rnu, CLEA Japan, Inc., Tokyo, Japan) aged 10–13 weeks were used for transplantation. MI model rats were created as previously described11. Rats whose hearts revealed less than 40% fractional shortening (FS) by echocardiogram just before MI induction were enrolled in further experiments. Cell sheet transplantation was performed 1 week after MI induction, which we call the “sub-acute phase” of MI. The sub-acute phase should be the optimal transplantation period that avoids damaging the transplanted cell with acute inflammation and obtains sufficient tissue repair before fibrotic scar formation in the chronic stage (Supplementary video 2)11,18. Rats were randomly divided into two groups: rats transplanted with cell sheets (Tx group) and sham-operated rats (sham group). The piling of 3-sheets and transplantation of cell sheets were performed as previously described11.
Transthoracic echocardiogram was performed with a Vivid 7 system (GE Healthcare, Waukesha, USA) and 11-MHz imaging transducer (GE 10S ultrasound probe; GE Healthcare, Little Chalfont, UK). Evaluations were performed as previously described11.
Histological analysis
Tissue sheets were fixed in 4% PFA and embedded with paraffin. Sections (6 μm thickness) were prepared and stained with Hematoxylin-Eosin and Sirius red. Cell sheets generated from the 836B3 human iPSC line were used for staining. Hearts were fixed in 4% PFA and embedded in OCT compound (Sakura Finetek Japan, Tokyo, Japan) and frozen. Five sections with 6-μm thickness were made at 50-μm intervals along the short axis from the center of the MI area for each rat and examined for Sirius red staining and immunofluorescence staining. MI length of fibrotic lesions at the level of the endocardium and total length of the endocardium were manually traced and measured with Sirius red staining sections and the ratio of the MI length and total length was calculated for each section. Values corresponding to each rat are an average of 5 calculated sections for each rat heart. For immunofluorescence staining, sections were treated with Protein Block Serum Free (DAKO) and incubated for 60 min with primary antibodies at room temperature. Anti-mouse Alexa 546 or 488 (1:500) and anti-rabbit Alexa 488 (Invitrogen) (1:400) were used as secondary antibodies. The area of engrafted human cells was manually selected as positive cell clusters for staining with human nuclear antibody (HNA) (mouse monoclonal, clone 235-1; Millipore, Billerica, USA) (1:100). The selected engrafted area and MI area (manually selected as lower panel of Fig. 4b) were measured and the ratio of the areas was calculated. Values corresponding to each rat are an average of 5 calculated sections for each rat heart. Anti-cTnT antibody (rabbit polyclonal; Abcam, Cambridge, UK) (1:500) was used for double staining with cTnT and HNA. For capillary density (capillary number/mm2), 20 random views of every rat heart that evenly included the border zone and central zone of the MI (×400, original magnification) were selected from vWF-stained sections and the number of capillaries was manually counted in each view. For vWF staining, a rabbit polyclonal antibody (DAKO) was used as the primary antibody (1:800). A mouse monoclonal cTnT antibody (clone 13211, Thermo Fisher Scientific) (1:200) was used for double staining with cTnT and vWF. All immunostained sections were photographed, measured and calculated with Biorevo BZ-9000.
Statistical analysis
The data were processed using Dr SPSS II software for windows (version 11.0.1J, SPSS Inc, Chicago, USA). Comparisons between two groups were made with the unpaired t-test. Comparisons between >2 groups were made with two-way analysis of variance (ANOVA) followed by Tukey's test as post hoc. Values are shown as mean ± SD. P values <0.05 were considered significant.
References
World Health Organization (WHO), The global burden of disease: 2004 update.http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_full.pdf (2008) (Date of access: 30/06/2014).
Ford, E. S. & Capewell, S. Proportion of the Decline in Cardiovascular Mortality Disease due to Prevention Versus Treatment: Public Health Versus Clinical Care. Annu Rev Public Health 32, 5–22 (2011).
Committee for Scientific, A., Sakata, R., Fujii, Y. & Kuwano, H. Thoracic and cardiovascular surgery in Japan during 2009: annual report by the Japanese Association for Thoracic Surgery. Gen Thorac Cardiovasc Surg 59, 636–667 (2011).
Anversa, P., Kajstura, J., Rota, M. & Leri, A. Regenerating new heart with stem cells. J Clin Invest 123, 62–70 (2013).
Joggerst, S. J. & Hatzopoulos, A. K. Stem cell therapy for cardiac repair: benefits and barriers. Expert Rev Mol Med 11, e20 (2009).
Masumoto, H. & Sakata, R. Cardiovascular surgery for realization of regenerative medicine. Gen Thorac Cardiovasc Surg 60, 744–755 (2012).
Thomson, J. A. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 282, 1145–1147 (1998).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Muller-Ehmsen, J. et al. Survival and development of neonatal rat cardiomyocytes transplanted into adult myocardium. J Mol Cell Cardiol 34, 107–116 (2002).
Teng, C. J., Luo, J., Chiu, R. C. J. & Shum-Tim, D. Massive mechanical loss of microspheres with direct intramyocardial injection in the beating heart: Implications for cellular cardiomyoplasty. J Thorac Cardiovasc Surg 132, 628–632 (2006).
Masumoto, H. et al. Pluripotent stem cell-engineered cell sheets reassembled with defined cardiovascular populations ameliorate reduction in infarct heart function through cardiomyocyte-mediated neovascularization. Stem Cells 30, 1196–1205 (2012).
Okano, T., Yamada, N., Sakai, H. & Sakurai, Y. A Novel Recovery-System for Cultured-Cells Using Plasma-Treated Polystyrene Dishes Grafted with Poly(N-Isopropylacrylamide). J Biomed Mater Res 27, 1243–1251 (1993).
Matsuura, K. et al. Transplantation of cardiac progenitor cells ameliorates cardiac dysfunction after myocardial infarction in mice. J Clin Invest 119, 2204–2217 (2009).
Yamashita, J. et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408, 92–96 (2000).
Yamashita, J. K. et al. Prospective identification of cardiac progenitors by a novel single cell-based cardiomyocyte induction. FASEB J 19, 1534–+ (2005).
Narazaki, G. et al. Directed and systematic differentiation of cardiovascular cells from mouse induced pluripotent stem cells. Circulation 118, 498–506 (2008).
Uosaki, H. et al. Efficient and Scalable Purification of Cardiomyocytes from Human Embryonic and Induced Pluripotent Stem Cells by VCAM1 Surface Expression. PLoS One 6 (2011).
Li, R. K., Mickle, D. A., Weisel, R. D., Rao, V. & Jia, Z. Q. Optimal time for cardiomyocyte transplantation to maximize myocardial function after left ventricular injury. Ann Thorac Surg 72, 1957–1963 (2001).
Sakakibara, Y. et al. Combined procedure of surgical repair and cell transplantation for left ventricular aneurysm: An experimental study. Circulation 106, I193–I197 (2002).
Kawamura, M. et al. Feasibility, safety and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation 126, S29–37 (2012).
Kawamura, M. et al. Enhanced survival of transplanted human induced pluripotent stem cell-derived cardiomyocytes by the combination of cell sheets with the pedicled omental flap technique in a porcine heart. Circulation 128, S87–94 (2013).
Madden, L. R. et al. Proangiogenic scaffolds as functional templates for cardiac tissue engineering. Proc Natl Acad Sci U S A 107, 15211–15216 (2010).
Don, C. W. & Murry, C. E. Improving survival and efficacy of pluripotent stem cell-derived cardiac grafts. J Cell Mol Med (2013).
Sakaguchi, K. et al. In vitro engineering of vascularized tissue surrogates. Sci Rep 3, 1316 (2013).
Shiba, Y. et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature 489, 322–325 (2012).
Lee, A. S., Tang, C., Rao, M. S., Weissman, I. L. & Wu, J. C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med 19, 998–1004 (2013).
Okita, K. et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 8, 409–412 (2011).
Koyanagi-Aoi, M. et al. Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proc Natl Acad Sci U S A (2013).
Laflamme, M. A. et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol 25, 1015–1024 (2007).
Acknowledgements
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labor and Welfare, Japan (to H.M., R.S. and J.K.Y.), the Project for Realization of Regenerative Medicine (to J.K.Y.) and Invited Research Project of Institute for Advancement of Clinical and Translational Science, Kyoto University Hospital (to A.M. and R.S.). We thank Dr. M.A. Laflamme (University of Washington) and Dr. H. Uosaki (Johns Hopkins University) for the detailed protocol of cardiac differentiation, Dr. K. Okita (CiRA) for the human iPSC line (836B3), Dr. B.B. Keller and Dr. W.J. Kowalski (University of Louisville) and Dr. P. Karagiannis (CiRA) for critical reading of the manuscript and Ms. F. Kataoka (Kyoto University) for technical assistance.
Author information
Authors and Affiliations
Contributions
H.M. designed and conducted the experiments, analyzed the data and wrote the main manuscript text. T.I., M.T. and H.F. conducted the experiments. A.M., T.I. and R.S. supervised the animal experiments. S.K. prepared figures 1a and 2a. T.S. and T.O. supervised cell sheet experiments. J.K.Y. wrote the main manuscript text and supervised this project. All authors reviewed the manuscript.
Ethics declarations
Competing interests
Yes there is potential Competing Interest. T. Okano is a director of the board of CellSeed Inc. T. Okano and T. Shimizu are stake holders of CellSeed Inc. Tokyo Women's Medical University is receiving research fund from CellSeed Inc. J.K. Yamashita is a founder, stake holder and scientific adviser of iHeart Japan Corporation and an inventor of pluripotent stem cell-related patents.
Electronic supplementary material
Supplementary Information
Supplementary video S1
Supplementary Information
Supplementary video S2
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Masumoto, H., Ikuno, T., Takeda, M. et al. Human iPS cell-engineered cardiac tissue sheets with cardiomyocytes and vascular cells for cardiac regeneration. Sci Rep 4, 6716 (2014). https://doi.org/10.1038/srep06716
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06716
This article is cited by
-
The Progress of Stem Cell Therapy in Myocardial-Infarcted Heart Regeneration: Cell Sheet Technology
Tissue Engineering and Regenerative Medicine (2022)
-
Application of mesenchymal stem cell sheet to treatment of ischemic heart disease
Stem Cell Research & Therapy (2021)
-
Possible NK cell-mediated immune responses against iPSC-derived cells in allogeneic transplantation settings
Inflammation and Regeneration (2021)
-
Scaffold-free cell-based tissue engineering therapies: advances, shortfalls and forecast
npj Regenerative Medicine (2021)
-
Targeted delivery of therapeutic agents to the heart
Nature Reviews Cardiology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
