
Abstract
Assembly of metagenomic samples is a very complex process, with algorithms designed to address sequencing platform-specific issues, (read length, data volume, and/or community complexity), while also faced with genomes that differ greatly in nucleotide compositional biases and in abundance. To address these issues, we have developed a post-assembly process: MetaGenomic Assembly by Merging (MeGAMerge). We compare this process to the performance of several assemblers, using both real and in-silico generated samples of different community composition and complexity. MeGAMerge consistently outperforms individual assembly methods, producing larger contigs with an increased number of predicted genes, without replication of data. MeGAMerge contigs are supported by read mapping and contig alignment data, when using synthetically-derived and real metagenomic data, as well as by gene prediction analyses and similarity searches. MeGAMerge is a flexible method that generates improved metagenome assemblies, with the ability to accommodate upcoming sequencing platforms, as well as present and future assembly algorithms.
Similar content being viewed by others

Introduction
The rapidly evolving state of Next Generation Sequencing (NGS) data has led to the development of a multitude of de novo and reference-based assembly tools1. To deal with the sheer number of reads generated by today's high-throughput NGS platforms, many of these new assembly tools utilize “Kmers” (words of length K) and de Bruijn graphs, as the method of choice for generating assembled contiguous sequence fragments (contigs). Each assembler yields different results (contigs and associated information), with some capable of generating ordered contigs if mate pair libraries are available2. Additionally, the results of any assembler can vary when altering any of a number of parameters, such as Kmer size selection, expected coverage, coverage cutoff, edge trimming or other tool-specific options3.
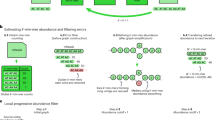
When a single genome is being sequenced, measuring the quality of the assembly is relatively straightforward, with a minimal number of large contigs being ideal. Large contig sizes, coupled with the fewest possible misassemblies, are always considered better. Increased sequencing of an isolate genome will eventually exhaust novel sequence information, as the entire genome is covered by an increasing number of reads; making sequencing and therefore assembly, a definable process. Metagenome assembly, in contrast, often has an unclear definition of assembly quality. Due to the number, frequency, types and sizes of genomes present in highly diverse communities, additional sequencing of metagenome samples often captures novel genome fragments from increasingly rare members (or minor variants) of the community. However, even when sequencing reaches or surpasses one terabase, novel reads continue to be generated when sequencing the most complex communities, such as those in soil4. This makes metagenomic sequencing and assembly difficult. There are at least two current approaches to metagenomic assembly, assembly of all data (typically requiring massive computational resources for raw assembly), or selection of a subset of the reads to assemble separately (binning or normalization). There are a number of issues with both methods, resulting in either poor contiguity despite the information being present in the raw data, or loss of data altogether. In addition, the methods are generally applied to raw data from a single sequencing platform. To mitigate these problems, we have designed an algorithmic approach called MetaGenomicAssembly by Merging (MEGAMerge) to recursively utilize a number of different Kmer based assemblies and combine the results into a single, merged contig set (Figure 1).

MeGAMerge pipeline for metagenomes.
This diagram provides an overview of the MeGAMerge process, including optional steps for trimming sequencing data and the inclusion of optional assemblers for Illumina reads. Long read or contig sets may include Sanger libraries, error-corrected PacBio reads (raw reads are likely to be too error-prone to be merged) and any other source of contigs. Input sequences of size < 200 bp are removed from this method, but this default value can be changed. The MeGAMerge pipeline currently uses Newbler to assemble short contigs and Minimus2 as the final assembly stage.
For metagenomes, contig N50 (size of contig where all contigs of equal or larger size add up to half the assembly size) and other traditional assembly metrics are less informative and frequently misrepresent the quality of the assembly. For such analysis, it is of value to include the total assembly size, which can include many very small contigs (which also shift the contig N50 and some other traditional statistics to inferior values). Measurements of large contig sizes remain a useful criterion for judging metagenomic assemblies; however assembly parameters or assemblers providing large contiguous fragments are rarely the ones that output the largest total assembly size. Additionally, as the size of the largest contig and total assembly are only two possible criteria for judging assembly, using these, or any other set of metrics for the selection of the “best” assembly or assembler for metagenomes may result in suboptimal performance. As there is currently no perfect tool for metagenome assembly, these assembly variations can be exploited by combining the results of different assemblies1,5,6.
To take advantage of the varied results generated when using different parameters and/or assemblers, MeGAMerge takes the resulting contigs from multiple assemblies and merges them together, coupled with long reads or other sequence data. The flexibility of this method allows the incorporation of any high-quality contigs into the process regardless of original source, making it adaptable to new technologies. The result is a single, merged assembly consisting of contigs of greater length and with better resolution than any single assembly. We demonstrate that this method consistently improves assembly metrics when applied to several different types of metagenomes. To validate that this procedure is applicable to a wide range of sequencing and assembly technologies, we analyzed MeGAMerge results for 21 metagenome samples with a minimum of 1 lane of Illumina sequence, some with additional 454 shotgun sequence data (Supplementary Table S1), as well as a synthetic dataset using synthetic illumine reads from 1000 reference genomes.
Since MeGAMerge is designed to combine various assemblies, including those from different assemblers, it can equally be applied to data from recently implemented, or upcoming, sequencing technologies (e.g. the long reads associated with Pacific Biosciences or the upcoming Oxford Nanopore technology), as well as data obtained from any assembly algorithm. Such a strategy allows the use of results from multiple assemblers, the application of multiple different parameters, as well as the inclusion of sequence data from multiple sequencing technologies. This method therefore accommodates future assembly procedures and novel technologies, to be included in the MeGAMerge process without significant alteration or updating of the methodology. Advances in technology, methodology or metagenomic theory will simply continue to improve metagenome assembly using this method.
Results
Comparison of Assembly Statistics
All MeGAMerge contigs show improvement of statistical metrics when compared with individual assemblies using the same assembler(s) within the same dataset. Table 1 illustrates the average improvements of assemblies broken down by data type. Supplementary Table S2 shows all metrics for all assemblies. Generally, MeGAMerge decreases the total number of contigs, by producing fewer, larger contigs. The total number of bases in MeGAMerge assemblies are comparable to the most bases assembled in the best single Kmer assembly, yet display greater contiguity (i.e. more bases contained in large (e.g. > 2 kb) contigs.
Of the five de novo Kmer based assemblers tested for this work (CLC Bio's denovo assembler (CLC Bio) and open source tools Velvet, SOAPdenovo, Ray and IDBA7,8,9,10), the two tools with metagenome implementations (IDBA and Ray) performed the best. CLC Bio performs well with respect to producing large numbers of assembled bases, but seems to assemble smaller contigs and show poorer support by read mapping. Individual assembly results for all metagenome samples examined, along with the results of merging contigs from one or more assemblies using our process (MeGAMerge) can be found in Supplementary Tables S2 and S3.
Analysis of “small” Kmer (< = 31) assemblies indicates that there does appear to be a trend of improvement of assemblies with increasing Kmer size from 21–31 (Supplementary Tables S2 and S3). It is not the case, however, that the largest Kmer (K = 31) contains all data represented in assemblies with Kmers 21–29. Additionally, use of larger Kmers (K>71) produces substantial improvements to the assembly (Supplementary Table S3). In fact, there is a consistent improvement of results when large Kmers, 454 data, or both, are utilized for MeGAMerge. Generally, when these data types are included, MeGAMerge produces a largest contig that is substantially larger than any produced in individual assemblies. This observation is likely due to the ability of larger Kmers or longer reads to span short repeats that shorter Kmers or reads cannot, coupled with the merging process that uses overlap-based consensus, rather than deBruijn graph based assembly, to join contigs.
IDBA, an iterative assembler conceptually similar to our method of merging results of multiple individual assemblies using MeGAMerge, also shows similar contig improvements. For example, use of IDBA with a restricted IDBA Kmer range (K = 21–31, steps of 2) provides results similar to MeGAMerge of individual assemblies with the same Kmer range (Supplementary Tables S2 and S3). Similar to observations of using larger Kmers with MeGAMerge (as stated above), improved assembly statistics resulted from the use of IDBA default assembly Kmer ranges (K = 20 to 100, by steps of 20). Additional improvements were observed when using the results of two or more assemblers as input to MeGAMerge, above and beyond the inclusion of multiple Kmers (see below).
Improvement to Assemblies using Small (K<31) Kmers
Results of representative small Kmer (K ≤ 31) assemblies from each tested assembler and from MeGAMerge results are shown in Table 1, as well as in Supplementary Tables S2, S3 and S4. For assemblies performed with Velvet, SOAPdenovo, or CLCBio, MeGAMerge produced improvements to the number of assembled bases, average contig size and in the number of bases contained in the largest contigs. Generally for small Kmers, the majority of assembly metrics show improvement as Kmer increases for metagenome samples (Supplementary Table S2). A consistent trend is seen in the reduction of the number of small contigs as well as the inclusion of more bases in the largest contigs of MeGAMerge assemblies (Supplementary Figure S1).
Using small Kmers, the largest MeGAMerge contig generated could be identical to a contig within one (or more) of the individual small Kmer assemblies. When this was not found to be the case, improvements to total contig length were often modest (Figure 2A). The presumptive reason for the minimal improvements in large contigs is the small Kmer size, which is unable to span and resolve low complexity, short repeats in the individual assemblies. However, alignments of all contigs from any individual assembly to the contigs produced by MeGAMerge indicates that not all parameters produce the largest fraction(s) of the largest MeGAMerge contigs (Supplementary Table S3).

MeGAMerge contigs encompass many smaller contigs from individual assemblies.
The largest MeGAMerge contigs produced using input assemblies from SOAP-denovo with Kmer ranges of 21–31 (A), or 85–99 (B), are shown for sample SRS022071. The underlying contig coverage is indicated with all contigs from the individual assemblies that are aligned to the MeGAMerge contigs (green and red lines indicate the 5′-3′ orientation of the original contigs). The read coverage is also shown using a sliding window of 100 bp. Both contig and read coverage support the MeGAMerge produced contigs.
Due to the nature of IDBA, it is not possible to perform MeGAMerge on IDBA output alone as this program already iterates over several Kmers to produce a single assembly. Contigs produced with IDBA (Kmers 21–31, by steps of 2) did produce improved contig statistics compared to any of the individual Kmer assemblies from Velvet, SOAPdenovo, or CLCBio (Table 1). However, MeGAMerge results, combining the same Kmer-range from these individual assemblers, outperformed this IDBA assembly.
The more recently available Ray assembler, was able to perform better with the HMP sample than the other assemblers and even produced better metrics than MeGAMerge contigs from other assemblers (with small Kmer assembly outputs). From read-mapping validation (see below), it appears that Ray also generates a class of contigs with notably higher fold coverage than other assemblers (Supplementary Figure S2). This improvement is not seen in the oil spill sample, where Ray performed approximately as well as several other assemblers. The variation in assemblers' ability to produce high quality assemblies illustrates the difficulty of selecting any particular assembler, or assembly, for the diverse array of possible metagenome samples. Applying MeGAMerge to multiple Ray assemblies produced considerable improvements in the number of bases assembled, the number of coding regions detected, as well as other assembly statistics (Table 2), indicating that even when Ray appears to be an individually superior algorithm for one particular sample type for metagenome assembly, MeGAMerge is able to improve upon any of these individual assemblies. The computational requirement for Ray is drastically higher than for any other assembler, however, potentially limiting its use with large datasets.
Longer Read Technology Helps Improve MeGAMerge Assemblies of Small Kmers
For small Kmers with all assemblers, MeGAMerge results are greatly improved by incorporation of a different technology, 454, which generates longer read data (Table 1). For example, MeGAMerge of Velvet contigs shows 8-fold increase in the size of the largest contig, with commensurate improvements in other categories when 454 reads are included into the inputs. Similar to what was conducted when using only small Kmer assemblies, the contigs from individual assemblies were aligned with the largest MeGAMerged contig obtained from the merged individual small Kmer and long read assemblies. In this case, the inclusion of longer reads generally results in novel large MeGAMerge contigs that are not represented in their entirety within any of the individual input assemblies. These contigs are supported both by the contig mapping results as well as read mapping-based validation, which is discussed below. The ability to include long reads into MeGAMerge natively allows the longer reads to join multiple contigs into a single contig. This parallels the observation that larger Kmers used for Illumina assemblies generate better metagenome assembly statistics than the use of smaller Kmers.
Long Contig Generation Improves When Using Large Kmers or Long Reads
Improvements to assembled contigs similar to those produced with 454 reads are also seen with Illumina-only assemblies when larger Kmers (85–105) are used as input to MeGAMerge (Table 2, Supplementary Table S3, Figure 2B). Furthermore, compared with individual Kmer assemblies, all contig statistics, including largest contig sizes, are improved once MeGAMerge is applied to these source datasets. The human microbiome sample SRS022071 was further examined as it contains both long (100 bp) Illumina reads and 454 data. Testing the incorporation of 454 reads together with a range of large Kmer assemblies using MeGAMerge did not produce substantial improvements, indicating that the observed differences are primarily due to read and Kmer length, rather than differential sequencing by platform. These improvements are likely the result of large Kmers mitigating many of the same assembly issues addressed by the longer 454 reads, as mentioned previously. The benefit observed, even when using Illumina reads alone with long Kmers, reiterate the universal need for longer reads, regardless of sequencing or assembly technology (assembly Kmer size is limited by read length).
MeGAMerge Improves Assembly when Merging Results from Multiple Assemblers
As discussed previously, IDBA using either a range of Kmers 21–31 or using default parameters produced contigs with metrics consistent with, or better than those produced by other individual Kmer assemblers. However, MeGAMerging of IDBA contigs with contigs from other assemblers consistently produces an improved assembly over IDBA alone (Table 1 and Supplementary Table S4). This supports the premise that individual algorithms are able to assemble different types of read-overlaps and produce different groups of contigs. Similarly, applying MeGAMerge to Ray assemblies in combination with results from other assemblers also yielded assembly improvements, including improvements over the merging of Ray contigs alone (Table 2 and Supplementary Table S4), indicating again that each assembler is capable of producing distinct contigs (possibly from different members of the community), even if the assemblies originate from the same read set(s) and using similar Kmer sizes to construct the graph. These results indicate that while assembly technology continues to progress, MeGAMerge can consistently improve upon the non-deterministic assemblies currently being produced and holds promise for continued success.
Sample Type Has Strong Effect on Assembly And MeGAMerge Improvement
The majority of the samples analyzed in this report are from human associated metagenomes and as such, are more likely to be relatively low in terms of sample complexity. This means that a much higher proportion of reads can be incorporated into these assemblies than for more complex metagenomes, such as available marine or soil metagenomes. When compared to a pelagic metagenome sample taken during the Horizon oil spill in the Gulf of Mexico, it can be seen that, there is a consistent improvement in assembly metrics when using MeGAMerge compared with individual Kmer based assemblies. Figure 3 shows the variation in two metrics of assembly (total bases in contigs larger than 2 Kb and average contig size in contigs > 2 Kb) for the Horizon oil spill metagenome and for a selected sample from the HMP (SRS022071) for comparison. It can be seen here that different samples and different assemblers produce highly variable results, however, MeGAMerge improvements are clear with either sample type.

Comparison of Statistical Metrics of Assembly for HMP and Oil Spill data.
Panel 3A shows the results of various assemblers compared to MeGAMerge for the average contig size (x-axis) and the total assembled bases (y-axis). MeGAMerge performs better than all other assemblers. Panel 3B shows the same graph for assembly of the oil spill sample. There is less uniformity for this sample, but MeGAMerge continues to produce more bases at a large average contig size.
Table 2 (and additional details in Supplementary Table S4) summarizes: 1) the use of different Kmer-based assemblers on metagenomes with Illumina reads; 2) the value of MeGAMerging the assembly results of any one assembler with multiple different parameters (varying Kmer sizes); and 3) the improvement when combining various assemblers and/or data types (Illumina and 454) with MeGAMerge.
Read Coverage Validation of Assembly
Read based validation of assemblies of the metagenome samples reveals what appears to be assembler specific characteristics, while validating the majority of assembled and MeGAMerge produced contigs. Figure 4 shows the results of read mapping to assemblies of a single Kmer and MeGAMerge of assemblies for both an HMP and the oil spill datasets (MetaHIT example in Supplementary Figure S3). Larger contigs with near complete read coverage can be observed. The lower coverage of small contigs is in part an artifact of read-mapping edge effects.

Read-mapping validation of HMP and Oil Spill produced contigs.
Percent coverage (y-axis) versus size of contig (x-axis) for MeGAMerge (black) and a single Ray Assembly (red) are displayed for HMP sample SRS022071 (A) and the oil spill sample (B). MeGAMerge contigs follow a similar pattern as with Ray contigs, with larger contigs that are validated by reads.
The majority of the variation and read-mapping improvements shown by MeGAMerge for these samples are due to improvement in the overall number of bases in large contigs, rather than introduction of novel data (Table 2). This can be seen much more clearly in the oil spill sample, where MeGAMerge-produced contigs are able to recruit more total reads than the number of reads recruited in all input assemblies (Supplementary Tables S5 and S6).
The relatively low complexity of the HMP samples results in a much higher percentage of read incorporation for all assemblies, with less of a distinct improvement upon merging assemblies with MeGAMerge. The microbial diversity of metagenome from the oil spill sample is much higher and shows a much greater degree of variability between assemblers and a more distinct improvement after using MeGAMerge. The proportion of reads recruited to contigs for the oil spill sample range by more than two orders of magnitude. For this sample, CLC bio produced the second most bases in contigs > 2 Kb, but read-mapping indicates that a much lower proportion of these contigs are recruiting unique reads. Additionally, the average contig coverage of individual CLC bio assemblies is much lower (95%) than expected (> = 99%) and the resulting MeGAMerge contigs are also substantially less well covered (85%). When allowing multiple alignments per read, rather than restricting to only one per read, improves this number to 88.2%, which is still much lower than any other assembler, putting to question the ability of CLC bio to properly assemble complex metagenome samples. When MeGAMerge is applied to all assemblies of this sample with the exception of CLC bio, the read-mapping based validation looks highly similar to the HMP results, with read-mapping again reaching 99% contig coverage and a similar size in the total assembled contigs. While IDBA behaves comparably to MeGAMerge applied to ranges of Kmer assemblies from other assemblers when using HMP samples, it performs less well on the more complex oil spill sample.
These results indicate that read-mapping is a vital tool for assessment of assemblies and that there is no uniformly ideal assembler for different sample types, lending further support to the utility of a method such as MeGAMerge, which can capitalize on the strengths of multiple assemblers to produce an improved final assembly.
Gene Prediction Supports Improvement by MeGAMerge
Analysis of the predicted coding regions from the contigs from individual assemblies and of MeGAMerge results indicates that coding regions are found in the majority of assembled contigs. MeGAMerge coding density is comparable or even improved when compared to individual assembly tools and this coding density (~90%) falls within the range of what is expected for microbial genomes. There is a consistent increase in the total number of predicted genes in MeGAMerge contigs when compared to individual Kmer assemblies of the same data. The increase in the number of predicted genes and the overall increase in average length in MeGAMerge contigs indicates that not only can more genes be predicted, but that more of these predicted genes will be found on large contigs, allowing for the analysis of linked genes (gene neighborhoods) and their associated functions, a critical element in metabolic pathway reconstruction.
Validation with Contig Alignments, Read Mapping and Similarity Searches
To further validate the MeGAMerge process and its ability to produce accurate larger contigs, alignments of input contigs and reads are performed with the output MeGAMerge contigs as part of the process. In Figure 2, we provide an example where the largest contig produced by MeGAMerge from HMP sample SRS022071, under two input assembly parameters (SOAPdenovo, Kmer ranges 21–31 and 85–99) were examined in depth. The results of mapping the input contigs from the original assemblies show that for the small Kmer range (21–31), the overlaps are normally long and in this case, there are three input contigs that can cover the entire length of the contig (Figure 2A). In contrast, the largest contig produced by MeGAMerge of contigs produced by large Kmer assemblies (85–99) exhibits a different behavior, with many additional contig joins and small contigs providing joins between larger contigs (Figure 2B). In both cases, there is a wide variation of the number and size of contigs that overlap with one another. Additionally, the overlaps between contigs range from near the minimal allowed overlap (of 80 bp), to thousands of bases.
As expected, read-based coverage across these contigs is variable and regions of lowest coverage are not necessarily found within the regions where contigs are joined, indicating strongly that MeGAMerge is producing a valid contig that is simply not present in any of the input assemblies. These regions of lowest read mapping coverage are widely distributed across the contig and are frequently in the center of contigs produced by the initial assembly, indicating that if errors are present in the original assemblies, they may be propagated by MeGAMerge, but do not appear to arise because of the MeGAMerge process.
To further provide support for the contiguity of these long MeGAMerge contigs, comparisons of these two contigs with other genomes was performed. It is important to note that this type of homology-based validation can only be performed if a sufficiently similar genome exists as an entry within the database. In this case, both largest contigs when aligned to NT were found to be highly similar to Bacteroides, known dominant gut microflora. The largest contig derived from the small Kmer range was aligned to its best hit, Bacteroides helcogenes P 36–108. This 342.5 kb contig was colinear along its entire length with a portion of the B. helcogenes chromosome, with a number of gaps where genomic islands or otherwise dissimilar regions intervened (Supplementary Figure S4). For the large Kmer contig, complete alignments to the best hits in NR were not possible as these were to genomic sections of uncultured organisms whose entries in NR were smaller than the MeGAMerge contig. However, alignments to the four next best completed or drafted genomes displayed gross similarity throughout the bulk of the 472.5 kb long contig (Supplementary Figure S5). Although a number of rearrangements can be observed between the MeGAMerge contig and each of the genomes, many rearrangements also exist between the genomes themselves. These results support MeGAMerge as a useful tool in providing longer contiguous contigs, without introducing any obvious chimeras.
Validation and chimera evaluation using synthetic data
While the above data support Megamerge as producing longer contiguous fragments matching known genomes, there is a risk that chimeras or other erroneous contigs are introduced into the final assembly. In order to investigate this, an Illumina read set representing a synthetic community of 1,000 genomes was generated. The data were assembled using multiple Kmers and also using Megamerge. All contigs were aligned to the original references to identify chimeras, to find contigs that did not map to the references and to calculate reference genome coverage. The individual Kmer assemblies (Kmers 51 to 71) resulted in contigs that covered from 41% to 73% of the reference genomes (Supplementary Table S7). The number of contigs from all individual Kmer assemblies combined totaled 11.7 million after dereplication and together, covered 80.26% of the references, with 318 contigs unable to be mapped to any reference. The MeGAMerge process reduced the total number of contigs to <3.7 million, covering 80.25% of the references, with only 114 contigs unable to be mapped to the references.
The number of chimeras present within each individual Kmer assembly increased with decreasing Kmer size and ranged from 491 to 3,651 per assembly. After dereplication (see Supplementary Figure S6 for the effect of dereplication), 10,087 chimeras remained and all contigs were used as input for MeGAMerge. The MeGAMerge process reduced the total number to 9,225 chimeras, of which 6,940 were carried over from the original contigs. Because several of the 1,000 genomes selected belonged to the same species (Supplementary Table S8), we further investigated if the chimeras (both new and carried over) were the result of merging non-contiguous regions within the same genome, or if they were generated among different genomes. We also investigated if the chimeras overlapped at repetitive regions within the genomes. Compared with the chimeras carried over from the individual Kmer assemblies, the MeGAMerge process has a higher proportion of chimeras generated within a genome (Supplementary Figure S7). The proportion of MeGAMerge chimeras found within repetitive regions is also superior to the proportion of the chimeras carried over from the individual assemblies.
Discussion
Here we present MeGAMerge, a method to generate improved metagenome assemblies by merging suboptimal assemblies of next generation sequencing data and which results in accurate and larger, more contiguous sequences. This method has already been used to successfully improve many assemblies of metagenome samples11,12,13,14. The speed of Kmer/de Bruijn-based assemblers has made these methods most practical for metagenome assembly, given the current state of sequencing, bioinformatics and computational technologies. MeGAMerge consistently improves almost all assembly metrics examined for a wide variety of metagenome datasets, regardless of data type, assembler, or parameter variation (Table 2). The observed improvements are highly interesting, as they indicate that the current tools used for metagenomic assembly do not make full use of the available data for contig construction regardless of parameter settings. Analysis of the MeGAMerge method has shown that longer read data and use of larger Kmers for assembly, can have a dramatic effect on the resulting MeGAMerged contigs.
There are known limitations to the number of contigs that can be merged in this manner: namely, the tools currently in use (see Methods), overlap based consensus tools' run-times increase exponentially with increasing input dataset size. However, better clustering, improved methods overlap consensus based assembly and binning of contigs into overlapping groups are all currently being explored as methods to improve and streamline this strategy.
As part of the MeGAMerge process, very small contigs are removed from the dataset, in order to ignore poorly covered, difficult to assemble, or other error-prone data from the individual assemblies, which also helps reduce the run time of MeGAMerge. This removal does not greatly impact any comparative metric, other than those related to addition of contig number or contig sizes, including total assembly size (and N50/N90), which are not very useful for comparing metagenome assembly performance. It is always the case that the largest MeGAMerge contigs contain more bases than an equal number of largest contigs from any individual input data set (i.e. fewer contigs are required to reach a certain number of bases). This indicates that individual assemblies contain unique sequence contiguity at each Kmer and generate contigs that overlap, in part or full, with those in other assemblies (Figure 2).
We have validated the contigs produced from the MeGAMerge process to be highly accurate based on read and contig mapping results. Furthermore, using simulated data for 1,000 genomes, we show that MeGAMerge greatly reduces the total number of (already dereplicated) contigs originally input into MeGAMerge, while maintaining reference genome coverage. In addition to this improved contiguity, MeGAMerge removes a large number of contigs that could not be mapped to the references (i.e. removes incorrect contigs) and while the merging process does introduce new chimeras, MeGAMerge reduces the total number of chimeras input into the process.
MeGAMerge contigs can be produced from any set of related sequence data (typically, but not necessarily restricted to assemblies and reads generated from the same sample) and results in more contiguous assemblies regardless of the original tool(s) used to generate the input assembly datasets. The use of MeGAMerge to allow merging of contigs from similar samples (environments) is possible and may theoretically allow improved reference contigs for large studies with many similar samples.
Perhaps most importantly, MeGAMerge is agnostic of the source of original data (platform), of the assembler(s) and of the parameters used to generate contigs, allowing the merging of two or more related assemblies or read sets, including those generated from older Sanger sequencing or newer Ion Torrent/Proton, or even high quality PacBio RS data. This method is therefore a powerful novel and enduring strategy for improving metagenomic assemblies, with the potential to continue to enable high quality metagenome assembly despite continuous changes in sequencing chemistries, platform upgrades, or even technology revolutions, in addition to the constant introduction of new assembly and clustering tools.
Methods
Samples
Supplementary Table S1 shows all 21 metagenome data sets used for these experiments, including read length, number of reads and total bases included in each sample, including number and types of reads, as well as average read length for Illumina reads. Briefly, 21 available metagenome samples were selected for this study: 3 from the MetaHit group15 (MH0001, MH0024, MH0042), 17 shotgun sequencing samples from HMP, including 3 samples with both Illumina and 454 data and Illumina data gathered during the Horizon oil spill. Average Illumina read lengths for these samples varied from 44–150 bp. Additional samples from the Human Microbiome Project (HMP) are also included.
Synthetic reads were generated using MetaSim and a customized Illumina error model, with per-base quality assignments provided based on data derived in-house. A total number of reads similar to a single Illumina HiSeq 2000 lane were generated for 1000 randomly selected genomes (see Supplementary Table S8), with an even distribution of input members. Each dataset consisted of 300 million (M) 100-bp, paired-end reads.
Assembly Methodology
Reads were trimmed using a sliding window based trimming protocol, with a minimum quality score of 2 to remove data of poor quality. For the purposes of this paper, SOAPdenovo16, as well as CLC Bio de novo assembler and Velvet7, were used for all individual Illumina assemblies, with a range of Kmers, typically from 21–31, for the data described here and using the ranges from 45–105 for samples with longer Illumina reads. Additionally, the more recently released assemblers IDBA and Ray were tested for several samples. If 454 reads were available, the Newbler assembly tool (454 Life Sciences, Roche) was also used to generate assembly of these reads independently. Assembled contigs are used for the merging steps described below. Unless otherwise noted, all assemblers were run with default parameters, utilizing samples as paired-end reads where applicable.
Kmer size is one of the most important parameters in short read assemblers and as read length increases for Illumina, larger Kmers can be used for assembly. Since the read sets available for this publication and used in this paper vary in length from 40 to 100 bp, our initial work was performed consistently with a range of Kmers from 21–31 for all assemblies. Additional validation of selected 100–150 bp data from the SRA was performed by merging assemblies with Kmers 85–105. This was performed primarily to investigate the hypothesis that assemblies show greater improvement in metrics when merging assemblies performed with longer Kmers.
MeGAMerge Methodology
Several tools are used in succession to combine the results of multiple assemblies/assemblers into a final contig set. All generated contigs are pooled and dereplicated to remove exact duplicates of contigs (or contigs that are subsets) arising from the multiple, different individual assemblies. The effect of dereplication can be seen in Supplementary Figure S6. Dereplication of contigs is performed using a custom implemented Perl script, to remove exact duplicates, reverse compliments and subsequences. Finally, contigs are binned together based on a size cutoff of 2.0 kb. Smaller contigs are assembled with Newbler and this output is combined with the pool of contigs greater than 2.0 kb. Finally, the sequences generated by Newbler and the original contigs greater than 2.0 kb in length are MeGAMerged using Minimus2, a tool from the AMOS toolbox17,18. Minimus2 has been modified to allow for ambiguous bases to be included in the assembly. Additional input assemblies using the CLC bio de novo assembler and Velvet7 were also tested and the pipeline was validated with similar results. A flowchart of the methodology is shown in Figure 1. The software is freely available at https://github.com/LANL-Bioinformatics/MeGAMerge.
Validation of Assemblies and MeGAMerge Contigs
There are multiple methods for measuring the success of a de novo assembly. These are typically statistical measures of the length of the assembled contigs. The following measures are frequently used or described as possible metrics for metagenome assembly: size of the largest contig, number of bases/contigs within a size category above an arbitrary cutoff (e.g. contigs above 10 kb in size), number of bases in the largest contigs (e.g. top 10, or 100 contigs), or number of contigs required to reach a defined set of bases (e.g. million). Any one of these metrics only provides a snapshot of a given assembly and thus these must be examined holistically to determine the success of an assembly.
Additional statistics are gathered by mapping reads to final MeGAMerged contigs using BWA19. The resulting BAM file was parsed using SAMtools to provide other metrics including percentage of each contig covered by reads and average fold coverage of each base of each contig. Figures were generated using the R statistical package. For further validation of assembly of the large contigs, Bowtie220 was used to map initial SOAPdenovo and Newbler contigs to the 100 largest merged contigs.
Reference genome coverage was obtained by aligning contigs to the genome sequences using Nucmer. For the discovery of chimeric locations using synthetic datasets, the resulting contigs were mapped to the references using BWA mem. Contigs that did not contiguously align to a single reference genome were flagged and the locations of the chimeras were identified when any single contig mapped to more than one reference genome. The chimeric locations located in repeat regions and at circular genomes' start/end positions were identified. Based on the reference genomes' gi numbers, the taxonomy compositions of chimeric sequences were analyzed to identify if the chimeras were among strain or species near neighbors.
Gene Prediction
The extent of assembled metagenomic contigs to be annotated was examined by use of the gene prediction tool Prodigal21,22. The types of statistical measures that can be generated using gene prediction include the number of predicted genes, as well as the predicted coding density of contigs. While these values are not expected to change much across assemblies, a substantial change coding density or in the ability of contigs to be annotated would be indicative of assembly quality.
For each assembly analyzed, contigs smaller than 2 kb were removed from the assembly. Contigs were then run through Prodigal Version 2.6 using the parameters (-c (closed ends) and -p meta) to produce GFF3 files. These files were then parsed to determine the size, number and coding density of predicted genes in all contigs.
References
Scholz, M. B. et al. Next generation sequencing and bioinformatic bottlenecks: the current state of metagenomic data analysis. Curr Opin Biotech 23, 9–15, 10.1016/j.copbio.2011.11.013 (2012).
Miller, J. R. et al. Assembly algorithms for next-generation sequencing data. Genomics 95, 315–327, 10.1016/j.ygeno.2010.03.001 (2010).
Earl, D. et al. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res 21, 2224–2241, 10.1101/gr.126599.111 (2011).
Pell, J. et al. Scaling metagenome sequence assembly with probabilistic de Bruijn graphs. P Natl Acad Sci USA 109, 13272–13277, 10.1073/pnas.1121464109 (2012).
Desai, N. et al. From genomics to metagenomics. Curr Opin Biotech 23, 72–76, 10.1016/j.copbio.2011.12.017 (2012).
Nijkamp, J. et al. Integrating genome assemblies with MAIA. Bioinformatics 26, i433–i439, 10.1093/bioinformatics/btq366 (2010).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829, 10.1101/gr.074492.107 (2008).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18, 10.1186/2047-217X-1-18 (2012).
Peng, Y. et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428, 10.1093/bioinformatics/bts174 (2012).
Boisvert, S. et al. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biology 13, R122, 10.1186/gb-2012-13-12-r122 (2012).
Mason, O. U. et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. Isme J 6, 1715–1727, 10.1038/ismej.2012.59 (2012).
Ishii, S. et al. A novel metatranscriptomic approach to identify gene expression dynamics during extracellular electron transfer. Nat Commun 4, 10.1038/Ncomms2615 (2013).
Dodsworth, J. A. et al. Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4, 1854, 10.1038/ncomms2884 (2013).
D'Haeseleer, P. et al. Proteogenomic Analysis of a Thermophilic Bacterial Consortium Adapted to Deconstruct Switchgrass. Plos One 8, e68465, 10.1371/journal.pone.0068465 (2013).
Qin, J. J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59-U70, 10.1038/Nature08821 (2010).
Li, R. Q. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20, 265–272, 10.1101/gr.097261.109 (2010).
Pop, M. et al. Comparative genome assembly. Brief Bioinform 5, 237–248, 10.1093/bib/5.3.237 (2004).
Sommer, D. D. et al. Minimus: a fast, lightweight genome assembler. Bmc Bioinformatics 8, 10.1186/1471-2105-8-64 (2007).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, 10.1093/bioinformatics/btp352 (2009).
Langmead, B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chapter 11, Unit 11 17, DOI:10.1002/0471250953.bi1107s32 (2010).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. Bmc Bioinformatics 11, 10.1186/1471-2105-11-119 (2010).
Hyatt, D. et al. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28, 2223–2230, 10.1093/bioinformatics/bts429 (2012).
Acknowledgements
Thank you to Paul Li for his work contributing to this paper, specifically for help in using Prodigal for validation of contigs. This study was supported in part by the U.S. Department of Energy Joint Genome Institute through the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and grants from the U.S. Department of Homeland Security under contract number HSHQDC08X00790 and the U.S. Defense Threat Reduction Agency's Joint Science and Technology Office (DTRA J9-CB/JSTO) under contract numbers B104153I and B084531I.
Author information
Authors and Affiliations
Contributions
M.S. designed the software and performed testing for this manuscript. C.C.L. was responsible for improvements to the code, including speed and functionality, as well as read-mapping validation and producing charts for publication. P.S.G.C. was responsible for initiation of the project and experimental design. All authors contributed to the analysis and writing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Figures S1-S7
Supplementary Information
Supplementary Tables S1-S8
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Scholz, M., Lo, CC. & Chain, P. Improved Assemblies Using a Source-Agnostic Pipeline for MetaGenomic Assembly by Merging (MeGAMerge) of Contigs. Sci Rep 4, 6480 (2014). https://doi.org/10.1038/srep06480
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06480
This article is cited by
-
Enhanced mercury phytoremediation by Pseudomonodictys pantanalensis sp. nov. A73 and Westerdykella aquatica P71
Brazilian Journal of Microbiology (2023)
-
Terabase-scale metagenome coassembly with MetaHipMer
Scientific Reports (2020)
-
Enteric Virome and Carcinogenesis in the Gut
Digestive Diseases and Sciences (2020)
-
Functional screening of a soil metagenome for DNA endonucleases by acquired resistance to bacteriophage infection
Molecular Biology Reports (2020)
-
Viral and metabolic controls on high rates of microbial sulfur and carbon cycling in wetland ecosystems
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
