
Abstract
The gut microbiota (GM) consists of resident commensals and transient microbes conveyed by the diet but little is known about the role of the latter on GM homeostasis. Here we show, by a conjunction of quantitative metagenomics, in silico genome reconstruction and metabolic modeling, that consumption of a fermented milk product containing dairy starters and Bifidobacterium animalis potentiates colonic short chain fatty acids production and decreases abundance of a pathobiont Bilophila wadsworthia compared to a milk product in subjects with irritable bowel syndrome (IBS, n = 28). The GM changes parallel improvement of IBS state, suggesting a role of the fermented milk bacteria in gut homeostasis. Our data challenge the view that microbes ingested with food have little impact on the human GM functioning and rather provide support for beneficial health effects.
Similar content being viewed by others

Introduction
The gut is inhabited by microbial communities that form an intimate and beneficial association with the host. It is an open microbial ecosystem composed of resident commensals that are continuously exposed to transient exogenous microbes originating from the diet. Many fermented foods and yoghurt in particular, contain high quantities of live bacteria, typically up to 109 CFU/g. These foods have been major contributors to the human diet since the Neolithic Era1, yet our modern understanding of the impact of food-ingested bacteria on our resident gut microbiome remains limited. To date, a majority of studies failed to identify significant modulations of the resident human gut microbiota upon consumption of fermented food2,3,4,5,6. This is likely due to the use of methods lacking sufficient phylogenetic resolution at the species level.
During the last two decades, human gut microbiota structure has increasingly been assessed by culture-independent methods based on 16S rRNA gene quantification or sequencing7,8,9,10,11. These methods can assess the overall structure of microbial communities but remain restricted in phylogenetic identification i) to the genus or species-subgroup levels and ii) by the availability of phylogenetically characterized 16S rDNA sequences in public databases. High throughput DNA sequencing-based approaches have emerged as a powerful alternative to study the complex microbial communities12. This deep sequencing permitted the construction of the first human gut microbiota gene catalog13. Subsequently, quantitative metagenomics has achieved species-level resolution, not only for previously known organisms but also for unknown species14,15,16. These technical and analytical advances provide the tools to explore the composition and metabolic capacity of the microbiota with an accuracy heretofore unattainable.
We deployed and extended these advances to identify gut microbial species modulated by a fermented milk product (FMP) in a 4-week intervention study in subjects suffering from Irritable Bowel Syndrome (IBS) with constipation (n = 28)18. IBS is a chronic functional disorder of the intestine with a prevalence of 10% to 25% and where 67% of diagnoses are for women17.
We aimed to use a species-level metagenomic approach to identify specific members of the gut microbiome modulated by the FMP. We found that the FMP potentiated the production of butyrate and others short chain fatty acids (SCFA) and decreased the levels of the opportunistic pathogen, Bilophila wadsworthia. These modifications occurred in parallel with an improvement of IBS symptoms18. Metabolic reconstruction of known and unknown species suggests potential cross-feeding involving the food-ingested bacteria and resident commensals that might be relevant for the overall gut homeostasis.
Results
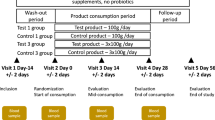
We administered a FMP or an acidified milk product (MP; 125 g/serving) to subjects fulfilling the Rome III criteria for IBS with constipation (IBS-C) during 4 weeks18 (Figure 1). Bacteria contained in the FMP were Bifidobacterium animalis subsp. lactis CNCM I-2494, Streptococcus thermophilus CNCM I-1630 Lactobacillus delbrueckii subsp. bulgaricus CNCM I-1632 and CNCM I-1519 and Lactococcus lactis CNCM I-1631. The consumption of the FMP improved the IBS condition of the subjects compared to the control group (i.e abdominal distension, acceleration of the oro-caecal and colonic transit times, overall IBS symptom severity)18. In order to assess the impact of the FMP on the gut microbiota of the enrolled subjects, we collected their stools before and after the intervention, extracted and sequenced their fecal DNA using a SOLiD v4 Next Generation Sequencing (NGS) device. We compared the sequencing data against the human gut microbiome catalog of 3.3 million genes13.

Study design and overview of the bioinformatic pipeline used in this study.
Genes of FMP species are detected in fecal samples
To validate the sensitivity of the method, we verified that DNA originating from the FMP bacterial species could be efficiently recovered in stool samples. We complemented the human microbiome catalog with 9030 non-redundant genes of the FMP genomes, otherwise absent, to insure an optimum mapping of the reads against the FMP genes. Abundance of all FMP species was significantly increased in stool samples in the FMP group at the end of the study. The low level signal detected at baseline and in control subjects is likely due to the presence of endogenous species phylogenetically close to the FMP species (Figure 2 A/B). B. lactis CNCM I-2494 was the dominant FMP species in stool, in agreement with its relatively high survival in the upper GI tract19,20.

The FMP and MP modulate species of the gut microbiota.
(A,B) Relative abundance of the FMP-species measured in FMP (A) and MP (B) groups at baseline and after intervention. (C,D) Relative abundance of known or unknown (MGS) species measured in FMP (C) or MP (D) groups at baseline and after intervention. Only MGS that were significantly modulated are depicted. Statistical significance is reported by asteriks (*;p<0.05, ***;p<0.001; Wilcoxon-paired test corrected for multiple tests comparison using the Benjamini-Hochberg procedure). Results are presented using Tukey box-and-whisker plots as quartiles (25%, median and 75%). Outliers were illustrated as single dots as per Tukey option of the Prism Graph software.
Endogenous microbial species are modulated by the FMP
Starting from a comprehensive gene-centric approach, we aimed to reduce data complexity from whole genome sequencing (WGS) reads to individual species. Our bioinformatics pipeline, summarized in figures 1 and S1, relied on 2 key principles: i) Increase (decrease) of a species results in enrichment (deprivation) of the abundance of its genes in the fecal microbiome. Consequently, gene counts should lead to identification of species modulated by the intervention. ii) Genes of a single species should have similar co-abundance profiles across individuals since they are physically linked on a DNA molecule (chromosome, plasmid, phage). As a result co-abundance based gene-clustering leads to identification of clusters of genes corresponding to one bacterial species as already demonstrated14,15.
We identified 1320 and 641 genes as significantly over- or under-represented after the consumption of the FMP or milk product (MP), respectively. We designated these FMP or MP “modulated genes”. Twenty seven percent of the FMP-modulated and 22% of the MP-modulated genes could be assigned to known species by blastN (Figure S2 A/B/C). Clustering by Spearman correlation was applied to the 1320 and 641 modulated genes using the MetaHIT cohort to increase statistical power and improve species assignment36.
Benchmarking against genes assigned to known species revealed that for clusters retrieved with a Spearman correlation factor of 0.75, 97% of genes were assigned to one cognate genome (i.e. known species) (Figure S3). This result confirms that clusters of genes identified can accurately serve as a proxy for intestinal bacterial species. Our analysis revealed that three clusters of modulated genes were derived from known species: Bilophila wadsworthia, Parabacteroides distasonis and Haemophilus parainfluenzae. Eleven additional clusters of genes without species-level identities were retrieved from the 1961 modulated genes (Figure S2D/E). The assignment of genes to unknown species allowed us to increase the percentage of genes without species-level identities for the FMP group from 27% to 51% (Figure S2A). Unknown species were assigned a number prefixed with the tag “MGS,” for MetaGenomic Species.
Integrating both known species and MGS results in a significant gain in statistical power through the compression of the number of variables as compared with the full catalog of 3.3 million genes. The identification of co-variant clusters also yields signatures that are more reliable than a single gene target. We computed the relative abundance of each known or unknown species by averaging the frequency of 50 representative genes for each species, as reported previously36 and compared the abundances using a Wilcoxon paired-test corrected for multiple testing. This revealed that consumption of the FMP stimulated MGS126, MGS203, MGS106, MGS109 and Bifidobacterium dentium. Three species, Parabacteroides distasonis, B. wadsworthia and Clostridium sp. HGF_2 were found to be inhibited (p<0.05) (Figure 2C). In the MP group, only two species, Haemophilus parainfluenzae and MGS204 varied significantly (p<0.05) (Figure 2D).
Reconstitution of the genetic repertoire of unknown species
We then attempted to extend the co-variance approach to the full catalog of 3.3 million genes in order to retrieve other genes of the FMP or MP modulated species. A unique gene of a given cluster was used as a “prey” to “chase” the genes of the catalog co-varying with the prey at a given threshold. Sets of co-varying genes from the full catalog were retrieved in a step by step approach and formed metagenomics clusters centered around the initial modulated genes identified above. We benchmarked this approach with 10 known species (Table S1) and retrieved an average of 2554 genes per species with a specificity of >95%, illustrating the applicability of this approach for retrieving genes of unique bacterial species (Table S1, cf. supplementary material). We applied the same procedure to the FMP-modulated MGS and retrieved >1900 genes for all the MGS (Figure S5A) but 2 (MGS106, MGS109), which had sub-bacteria-sized gene repertoires and were not further investigated. Five MGS were identified as Clostridiales including Eubacterium (MGS146), Clostridium (MGS134), Roseburia (MGS204) and unknown genera (MGS203, MGS126) and 2 MGS as Bifidobacterium (MGS109, MGS106) (Table 1, Supplementary Material).
The genetic repertoire of a MGS should contain the information needed to perform essential bacterial functions (e.g. DNA replication, peptidoglycan synthesis, protein synthesis). Essential genes represented an average 9.5% of MGS genetic repertoires, which is comparable to other gut commensals such as Escherichia coli and Bifidobacterium longum (Figure S5B). By the same token, it is expected that functions encoded by MGS should also cover the major metabolic pathways present in any microbial cell (i.e. carbohydrate, lipid, amino acids and nucleic acids metabolisms). Genes of MGS were assigned putative functions by comparing them to Cluster of Orthologous Groups (COGs)21 using the CD-Search Tool22. Sorting of these COGs by their hierarchical functional categories confirmed that functions encoded by the MGS similarly cover the major metabolic pathways compared to reference commensals (E. coli and B. longum) (Figure S5C). This result was supported by the projection of the MGS and E. coli on the global map of KEGG metabolism (Figure 3A, Figure S6).

Metabolic reconstruction of unknown gut microbial species shows a FMP-mediated increase of potential butyrate producers.
A) Projection on KEGG metabolic pathways of functions encoded by the MGS126 reconstructed genome (in red) using Ipath tool55. Functions of the KEGG global map were depicted underneath. B) Presence of genes predicted to encode enzymes of the butyrate synthesis pathway (thiolase EC 2.3.1.9 (THL); β-hydroxybutyryl-CoA dehydrogenase (BHCD; EC 1.1.1.35); crotonase (CRO; EC 4.2.1.17); butyryl-CoA dehydrogenase (BCD; EC 1.3.99.2); Electron-Transfer Flavoprotein α and β sub-units (ETFα and ETF β; E.C. 1.5.5.1); butyrate kinase (BK; EC 2.7.2.7); butyryl-coA acetyl-coA transferase (ButCoA; EC 2.8.3.8) among the reconstituted Clostridiales MGS. C) Abundance of butyrogenic modules of the human gut microbiota in the IBS cohort at baseline. Mean and standard deviation are represented. Results are presented using Tukey box-and-whisker plots.
We concluded that we retrieved nearly complete genomes of the FMP-modulated species, allowing further metabolic and ecological analyses.
FMP increases potential butyrate producers
The Clostridiales are primary butyrate producers. Our analysis revealed that MGS126, MGS203, MGS204 and Clostridium sp. HGF2 possess the genetic capacity to produce butyrate (Figure 3B).
In order to assess the impact of the FMP on the butyrate producing community, we reconstituted the butyrogenic modules of the entire human microbiome gene catalog by applying the gene-clustering algorithm to the genes of the catalog encoding the butyrate synthesis pathway (cf. Supplementary Methods). This procedure yielded 39 butyrogenic modules of which 30 could not be taxonomically assigned at the species level (Figure 3C). The 9 species-assigned modules were previously reported to belong to experimentally-confirmed butyrate producers Roseburia intestinalis, Roseburiainulinivorans, Butyriovibrio crossotus, Clostridium L2-50, Faecalibacterium prausnitzii, Eubacterium hallii, Lachnopsiraceae bacterium 5_1_63FAA, Coprococcus ART55/1 and Acidaminococcus intestini D2123,24,25. Among the 30 non-assigned species modules, 27 belong to the Order Clostridiales including 5 Lachnopsiraceae, 8 Clostridiaceae, 2 Veillonellaceae and 1 Bacteroidales. Each of these taxa is known to have members capable of butyrate production25,26,27,28. This result validated our approach, as we retrieved butyrogenic modules belonging to major known intestinal butyrate producers. The relative abundances of the butyrogenic modules were calculated across the studied cohort. Module 12, among the 10 most abundant, belongs to MGS126. Modules 44 and 20 belong to MGS203 and MGS204, respectively. As expected, in parallel with their respective MGS, modules 12 and 20 were significantly increased and decreased after FMP and MP consumption, respectively (Wilcoxon paired test; data not shown). A trend (p<0.10) showing an increase of the module 44 (MGS203) was also observed in the FMP group, although it did not reach statistical significance (data not shown). Intergroup comparisons of butyrogenic modules after the interventions indicated a higher abundance of module 4 (Roseburia inulinivorans) in the FMP group as compared to the MP group (p<0.05; ANCOVA without multiple test correction; p>0.05 after Benjamini-Hochberg multiple test correction; Figure S8).
Butyrogenic bacteria are known to produce propionate as an end product of L-fucose degradation29. The presence of L-fucose degrading genes in the genomes of MGS126 and MGS203 might indicate a metabolic potential to also produce propionate (Table 2).
FMP stimulates the production of SCFA in vitro
In order to further assess the impact of the FMP on short chain fatty acid production we used a 5-compartiment ex vivo human colonic fermenter (SHIME ® PRODIGEST)30 inoculated independently with 2 different healthy donors and kept stable for >5 weeks. We observed that the addition of the FMP led to a significant increase of butyrate, propionate and total SCFA, in the 3 different vessels mimicking the ascending, transverse and descending colon conditions as compared with baseline (Figure 4, Figure S7). Butyrate was the SCFA with the largest observed increased registering an average 1.9 fold augmentation across vessels and individuals (Figure 4).

The FMP stimulates the production of butyrate by human colonic microbiota in vitro.
The SHIME colonic fermenter mimicking conditions of the acending, transverse and descending colons (reference 30) was inoculated with fecal microbiota of 2 healthy human donors (black and grey columns) and after 2 week (C1 & C2 time points) exposed to FMP during 3 weeks (T1, T2 and T3 time points). Mean fold changes between control and treatment periods are reported when statistical significance was met. n.s: non-significant, *** (p<0.001, ANOVA).
Functions encoded by FMP-modulated bacteria
Next, we reasoned that species stimulated (or decreased) by the FMP might have responded to the same environmental intraluminal changes triggered by the ingestion of FMP bacteria. We aimed to have a better insight on habitat type, cell cycle and energy metabolism of the FMP modulated species by identifying shared functions of FMP-stimulated species absent in FMP-inhibited species and vice-versa. Available genome information of known species and the reconstituted genetic repertoires of MGS were used to perform comparative genomics and identify Clusters of Orthologous Genes (COGs)21 discriminating between FMP-stimulated and inhibited species (Table 2, TableS2).
Genes encoding pili were present in all FMP-modulated species indicating a potential for epithelial adhesion. MGS126 and MGS203 possessed enzymes allowing utilization of L-fucose which is a mucin derived monosaccharide. These 2 species also carried genes encoding the syntheses of flagellin and spores, which have both been reported to be involved in mucus colonization31,32. Taken together, this data informed us on the possible adaptation of these species to a mucous environment.
The presence of genes involved in oxidative phosphorylation (i.e. cytochrome oxidases bd) in B. wadsworthia and P. distasonis genomes indicates a shared capacity of these species to inhabit niches where oxygen tension is low (i.e. epithelial interface).
We further investigated whether inhibited species shared common features that would be absent in stimulated species. Genes encoding a putative carbon monoxide dehydrogenase (CODH) were found to be present in B. wadsworthia and Clostridium sp. HGF2 genomes predicting their capacity to use carbon monoxide (CO) as an electron donor33. Heme oxygenase is a known source of intestinal CO and its activity has been shown to be up-regulated during intestinal inflammation34. This suggests that CO utilization might be an advantage in inflamed conditions for organisms able to use it. B. wadsworthia also possesses the set of genes (i.e. HMP0179_00240-00243) allowing anaerobic respiration of nitrate, a substrate that is also enriched during inflammation35.
Discussion
Fermented foods have been a part of the human diet since ~10 000 BC comprising different matrixes such as milk (i.e kefir, yoghurt, dahi, cheese) or vegetables (i.e. nato, sauerkraut, pickles)1. Beneficial effects of fermented milks were empirically assessed by the Nobel Prize laureate Elie Metchnikoff in 1907 who predicted their role in the enhanced health and longevity of Bulgarian populations36. In the light of our increased understanding of the impact of the gut microbiome on host health, the question arises how food-ingested microbial communities modulate the autochtonous microbiota.
The emergence of molecular tools based on the 16S rDNA gene brought about a revolution in understanding the immense diversity of gut microbial communities and their links with host health and diet. However, variations of 16S rDNA sequences do not provide basis for species-level resolution8. Consequently, studies using 16S gene as a phylogenetic marker and reporting unaltered gut microbiota upon probiotic consumption2,3,4,5,6 might have failed in detecting changes of the microbiota due to the lack of species-level resolution. Such hypothesis can be accurately studied now with the emergence of novel technologies allowing species-level gut microbiota mapping.
In the current work, whole genome sequencing analysis coupled with a gene-centric approach was used to investigate the effect of a FMP on the human gut microbiota at the species-level resolution. Initially, only 27% (356 of 1320) of genes modulated by the FMP could be assigned to known species; illustrating the limited representation of bacterial commensals in publicly available genome databases. By employing gene clustering methodologies14,15,16, the species-level assignment of FMP-modulated genes was increased to 51%. This clustering algorithm was further used to reconstitute the bacterial gene repertoires of the unknown species used for comparative genomics.
Upon FMP consumption, two previously uncharacterized butyrate producers MGS203 and MGS126 were increased. MGS126 was among the 10 most abundant butyrogenic bacteria of the human gut microbiota. This argues for a positive effect of the FMP on the net production of butyrate in the colon. Importantly, this hypothesis was supported by our in vitro results obtained with the SHIME® fermenter and in a mouse model of intestinal inflammation37. MGS203 and MGS126 were likely stimulated by the FMP-mediated increase of Bifidobacterium species resulting in a higher production of acetate and lactate, used by butyrate producers23,38.
Recent studies have shown that B. wadsworthia and P. distasonis were increased in western diet enriched in saturated animal fat39,40 or RS4, a resistant starch used in processed foods41, respectively.
Although an unlikely potential effect of dietary changes between groups cannot be excluded to explain the decrease of these 2 species in the FMP group, we privileged the more likely hypothesis that the FMP modulated-species responded to FMP-mediated signals. We thus reasoned that species inhibited in the FMP group shared common metabolic pathways that we investigated by comparative genomics.
B. wadsworthia is a gram negative δ-Proteobacteria first isolated from an appendicitis patient and is often associated with inflamed and/or infected clinical samples42. Its genetic potential to utilize the known inflammation-associated metabolites like nitrate and possibly carbon monoxide, suggests that it can be fueled by host-derived products enriched during inflammation, similarly to other pathobionts (i.e. Salmonellaenterica)35. By the same token, the carbon monoxide dehydrogenase genes of another FMP-modulated species, Clostridiumsp. HGF_2, suggests that it could also take advantage of intestinal inflammation34. Both B. wadsworthia and Clostridiumsp. HGF_2 are decreased by the FMP, which could thus have an anti-inflammatory effect as observed in the mouse model of colitis37.
The FMP-induced changes in the GM paralleled improvement of IBS condition in our cohort, suggesting a role of the FMP-modulated species in the amelioration of the clinical parameters18. This hypothesis is compatible with the observations that i) butyrate, known to improve intestinal motility43,44 and visceral sensitivity45, is underproduced by IBS-C gut microbiota compared to healthy controls46 and ii) B. wadsworthia is pro-inflammatory40 and produces sulfide which is toxic47 and nociceptive48.
The MP has reduced effects on gut microbiota compared to the FMP which is consistent with the absence of an active microbial community in this product. Only 2 species, H. parainfluenzae and the Clostridiales MGS204, were shown to be modulated (i.e. decreased) upon the MP intervention. Changes observed in the MP group might originate from a natural variation of the gut community reported to be higher in IBS compared to healthy controls49. Alternatively, it is also possible that the MP induced changes in the gut microbiota. Since H. parainfluenzae is a γ-proteobacteria considered as a gut pathogen and previously associated with IBS in children50, MP-mediated changes might therefore account for the reported MP placebo effect18,51.
In conclusion, our study sheds light on the potential of the bacteria conveyed by fermented milks to stimulate synthesis of beneficial metabolites and decrease abundance of pathobionts. These modifications can potentially improve health and are thus of importance for public health recommendations in western countries. It indicates that the role of food-ingested bacteria in gut homeostasis has been under-estimated, possibly because of methodological limitations that can, today, be overcome. Elucidation of the intricate links between food ingested microbes and human symbionts can thus be addressed.
Methods
Subjects and study design
The study18, was a single centre, randomized, double-blind, controlled, parallel-group design including women (aged 20–69 years), fulfilling the Rome III criteria for IBS-C. Subjects were asked to not consume probiotic-containing products or fermented dairy products. There was no dietary record. After an 11-day wash-out period, 32 subjects (Per Protocol) consumed (125 g/serving) twice a day either the FMP (n = 17) or an acidified milk product (MP) (n = 15) for 4 weeks. Out of 64 stools collected within the PP population, 56 samples (n = 26 and 30, for FMP and MP, respectively) passed DNA quality control and were used for further analyses. Other dairy products and probiotics were excluded from the diet.
Study products
The FMP contained 1.25 × 1010 colony forming units per serving (cfu/serving) Bifidobacterium animalis subsp. lactis (strain number I-2494 in the French National Collection of Cultures of Micro-organisms (CNCM), Paris, France), 1.2 × 109 cfu/serving of Streptococcus thermophilus (CNCM I-1630) Lactobacillus delbrueckii subsp. bulgaricus (CNCM I-1632 and CNCM I-1519) and Lactococcus lactis (CNCM I-1631). The MP was an acidified milk product with low lactose content. FMP and MP were provided by Danone Research (Palaiseau, France).
Stool collection, storage, fecal DNA extraction and sequencing
Stool samples were collected before and after the 4-week consumption period. Immediately after defecation, a fecal sample was collected and stored in RNAlater solution (Ambion). The fecal suspension was homogenized and the volume of RNAlater was adjusted to achieve a final fecal dilution of 1:10 (wt/vol). 200 µl of the 10-fold dilution were added to 1 ml of Phosphate Buffered Saline (Sigma-Aldrich) and centrifuged for 5 min at 5,000 × g. The supernatant was discarded and the fecal pellet stored at −80°C. Fecal DNA was extracted as previously described52. DNA samples were sequenced on a SOLiD v4 NGS (Life Technologies) following the standard protocol for fragment libraries. The raw SOLiD read data was deposited in the EBI European Nucleotide Archive under the accession number PRJEB7171.
Identification of the species modulated by the interventions
Our bioinformatics pipeline, summarized in the Figure S1, aimed to decrease data complexity starting from reads and identify bacterial species modulated by the intervention. In brief, 50-bases tags (reads) were generated with SOLiD v4 sequencer, mapped on the MetaHIT 3.3 million genes catalog and genes count profiles were generated for each individual. Genes potentially modulated upon the intervention (intra-group analysis) were identified with a Kruskall-Wallis test, clustered and assigned to known or unknown species. Relative abundance of a given species was computed by averaging the frequency of 50 genes belonging to the species. DNA from the samples (n = 28) was sequenced with a SOLiD v4 sequencer and an average of 3.97 × 107 50-bases tags (reads) per sample were generated and mapped on the MetaHIT 3.3 million genes catalog13 (average 1.71 × 107 reads/sample; S.D. +/− 7.79 × 106) using METEOR, an in-house software53. Only those tags that mapped uniquely (univocally) to a single gene and to the exclusion of FMP genomes were retained (1.30 × 107 reads/sample; S.D. +/− 5.88 × 106). To quantify FMP species, the catalog of 3.3 million genes was complemented by the non-redundant genes (n = 9030) of the FMP genomes4,54. Genes count profiles were generated for each individual and genes significantly enriched or depleted upon the intervention (intra-group analysis) were identified with a Kruskall-Wallis test. Clusters of genes or species counting less than 9 (or 5) genes in the FMP (or MP) group were disregarded since these clusters are more likely to have resulted from random variations (see supplementary material).
Quantification of mapped reads to the annotated reference was used to identify genes belonging to the endogenous microbiota that were significantly modulated upon intervention. A Mann-Whitney test was performed for FMP and MP groups to identify genes over or under represented after intervention compared to baseline. These are subsequently referred to as “modulated genes.” 1320 and 641 modulated genes were identified for FMP and MP, respectively.
Spearman correlation coefficients were calculated for all gene pairs of this set, using gene abundance as a variable. Clusters of genes were defined as co-varying gene groups at 0.75 spearman correlation threshold. To augment the statistical power of the clustering analysis, we used the gene abundance matrix of 292 individuals from the MetaHIT consortium (Metahit cohort)15. Genes were assigned to a species at a threshold of 95% identity over 90% of the sequence by blastN14.
The genetic repertoire of unknown species was reconstituted through comparison of this study's gene clusters and the co-variation within the 3.3 million gene catalog across the MetaHIT cohort15. Functions for each clustered or reference genome genes were predicted using COG and/or NOG assignments. When available, COG assignment was performed using the Batch Web CD-Search online tool (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi). Predicted functions were projected on KEGG metabolic pathways using the Ipath software55 and compared to essential genes identified in the model organism Bacillus subtilis56. When species-level taxonomy could not be assigned, higher taxonomic levels were assigned using MEGAN57. Relative abundance was computed as the average frequency of 50 randomly selected genes of a species. Wilcoxon paired test, corrected for multiple tests by the Benjamini-Hochberg procedure, was used to test the significance of the FMP or MP effects.
Butyrate producers of the human gut microbiome
Genes involved in butyrate production from Roseburia intestinalis or Coprococcus eutactus25 were used as references to retrieve homologs present in the human gut microbiome catalog by using a bi-directional BlastP. Such genes were re-organized into co-varying clusters, hereafter called butyrate modules, which belong to unique species (cfsupplementary material). Relative abundance of the butyrate modules was calculated as the mean of the relative abundance of the genes composing the module. Wilcoxon paired test was used to test the statistical significance of the variation upon interventions (intragroup analysis). For intergroup comparisons, a one-way ANCOVA (Analysis of Co-Variance) was used on the change from baseline for each butyrate module with the group as factor. The relative abundance at baseline was considered as covariate. A Benjamini-Hochberg multiple testing correction was applied to control the False Discovery Rate.
FMP increases SCFA production in an in vitro colonic fermentation system
In vitro colonic fermentation model (SHIME® (Prodigest, Belgium)) mimicking the adult gastro-intestinal tract was used to assess the impact of FMP on production of gut microbial derived SCFA58. The system was inoculated with fecal sample from healthy adults (n = 2). After two-week stabilization period, 50 g of FMP was added daily to the stomach compartment during 3 weeks. The 3 vessels mimicking the ascending, transveral and descending colons were sampled 3 times a week. Concentrations of acetic acid, propionic acid, butyric acid were measured by liquid gas chromatography. A two-way ANOVA (Analysis of Variance) was used to evaluate the impact of FMP consumption on the production of SCFA. The first factor was the period of treatment and the second was the week of sampling nested in the period treatment. Tukey-Kramer post-hoc tests were used to determine which comparisons were significantly different if needed. Normality of residuals and homogeneity of variance were checked to validate ANOVA hypothesis. When the hypothesis of homogeneity of variance between groups was violated, the variance by week was used instead of a pooled variance to allow the use of ANOVA.
References
Prajapati, J. B. & Nair, B. M. The history of fermented foods in Handbook of Fermented Functional Foods. (ed. Farnworth, E. R), pp. 1–25. (CRC Press, Boca Raton New York, London, Washington DC, 2013).
Charbonneau, D., Gibb, R. D. & Quigley, E. M. M. Fecal excretion of Bifidobacterium infantis 35624 and changes in fecal microbiota after eight weeks of oral supplementation with encapsulated probiotic. Gut Microbes 4, 201–211 (2013).
Kajander, K. et al. Effects of multispecies probiotic supplementation on intestinal microbiota in irritable bowel syndrome. Aliment. Pharmacol. Ther. 26, 463–473 (2007).
McNulty, N. P. et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 3, 106ra106 (2011).
Michail, S. & Kenche, H. Gut microbiota is not modified by randomized, double-blind, placebo-controlled trial of VSL#3 in diarrhea-predominant irritable bowel syndrome. Probiotics Antimicrob. Proteins 3, 1–7 (2011).
Lahti, L. et al. Associations between the human intestinal microbiota, Lactobacillus rhamnosus GG and serum lipids indicated by integrated analysis of high-throughput profiling data. PeerJ 1, e32 (2013).
Smith, C. J. & Osborn, A. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67, 6–20 (2009).
Janda, J. & Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils and pitfalls. J. Clin. Microbiol. 45, 2761–2764 (2007).
Zoetendal, E., Rajilic-Stojanovic, M. & de Vos, W. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 57, 1605–1615 (2008).
Petrosino, J. F., Highlander, S., Luna, R. A., Gibbs, R. A. & Versalovic, J. Metagenomic pyrosequencing and microbial identification. Clinical Chemistry 55, 856–866 (2009).
Paliy, O. & Agans, R. Application of phylogenetic microarrays to interrogation of human microbiota. FEMS Microbiol. Ecol. 79, 2–11 (2012).
Lepage, P. et al. A metagenomic insight into our gut's microbiome. Gut 62, 146–158 (2013).
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010).
Qin, J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60 (2012).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013).
Karlsson, F. H. et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498, 99–103 (2013).
Canavan, C., West, J. & Card, T. The epidemiology of irritable bowel syndrome. Clin. Epidemiol. 6, 71 (2014).
Agrawal, A. et al. Clinical trial: the effects of a fermented milk product containing Bifidobacterium lactis DN-173-010 on abdominal distension and gastrointestinal transit in irritable bowel syndrome with constipation. Aliment. Pharmacol. Ther. 29, 104–114 (2008).
Rochet, V. et al. Survival of Bifidobacterium animalis DN-173 010 in the faecal microbiota after administration in lyophilised form or in fermented product - A randomised study in healthy adults. J. Mol. Microbiol. Biotechnol. 14, 128–136 (2008).
Collado, M. C., Moreno, Y., Cobo, J. M., Mateos, J. A. & Hernandez, M. Molecular detection of Bifidobacterium animalis DN-173010 in human feces during fermented milk administration. Food Res. Int. 39, 530–535 (2006).
Tatusov, R. L., Koonin, E. V. & Lipman, D. J. A genomic perspective on protein families. Science 278, 631–637 (1997).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2011).
Duncan, S. H., Louis, P. & Flint, H. J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 70, 5810–5817 (2004).
Jumas-Bilak, E. et al. Acidaminococcus intestini sp. nov., isolated from human clinical samples. J. Mol. Microbiol. Biotechnol. 57, 2314–2319 (2007).
Louis, P. & Flint, H. J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 294, 1–8 (2009).
Prabhu, R., Altman, E. & Eiteman, M. A. Lactate and acrylate metabolism by Megasphaera elsdenii under batch and steady-state conditions. Appl. Environ. Microbiol. 78, 8564–8570 (2012).
Goker, M. et al. Complete genome sequence of Odoribacter splanchnicus type strain (1651/6). Stand. Genomic Sci. 4, 200–209 (2011).
Stanton, T. B. et al. Recognition of two new species of intestinal spirochetes: Serpulina intermediasp. nov. and Serpulina murdochii sp. nov.. Int. J. Syst. Bacteriol. 47, 1007–1012 (1997).
Scott, K. P. et al. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the Human gut bacterium Roseburia inulinivorans. J. Bacteriol. 188, 4340–4349 (2006).
Van den Abbeele, P. et al. Microbial community development in a dynamic gut model is reproducible, colon region specific and selective for Bacteroidetes and Clostridium Cluster IX. Appl. Environ. Microbiol. 76, 5237–5246 (2010).
Lillehoj, E. P., Kim, B. T. & Kim, K. C. Identification of Pseudomonas aeruginosa flagellin as an adhesin for Muc1 mucin. Am. J. Physiol. -Lung C. 282, L751–L756 (2002).
Wiencek, K. M., Klapes, N. A. & Foegeding, P. M. Hydrophobicity of Bacillus and Clostridium spores. Appl. Environ. Microbiol. 56, 2600–2605 (1990).
Oelgeschlager, E. & Rother, M. Carbon monoxide-dependent energy metabolism in anaerobic bacteria and archaea. Arch. Microbiol. 190, 257–269 (2008).
Zhu, X., Fan, W. G., Li, D. P., Kung, H. & Lin, M. C. Heme oxygenase-1 system and gastrointestinal inflammation: A short review. World J. Gastroentero. 17, 4283–4288 (2011).
Winter, S. E., Lopez, C. A. & Baeumler, A. J. The dynamics of gut-associated microbial communities during inflammation. Embo Rep. 14, 319–327 (2013).
Metchnikoff, E. The Prolongation of Life: Optimistic Studies (G.P. Putnam's Sons, New York, NY, 1908).
Veiga, P. et al. A Bifidobacterium animalis subsp. lactis fermented milk product reduces inflammation by altering a niche for colitogenic microbes. P. Natl. Acad. Sci. USA 107, 18132–18137 (2010).
Duncan, S. H. et al. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 91, 915–923 (2004).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014).
Devkota, S. et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/-mice. Nature 487, 104–108 (2012).
Martinez, I., Kim, J., Duffy, P. R., Schlegel, V. L. & Walter, J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 5 (11), e15046 (2010).
Baron, E. J. Bilophila wadsworthia: a Unique Gram-negative Anaerobic Rod. Anaerobe 3, 83–86 (1997).
Soret, R. et al. Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterol. 138, 1772–1782 (2010).
Fukumoto, S. et al. Short-chain fatty acids stimulate colonic transit via intraluminal 5-HT release in rats. Am J. Physiol. -Reg. I. 284, R1269–R1276 (2003).
Vanhoutvin, S. A. L. W. et al. The effects of butyrate enemas on visceral perception in healthy volunteers. Neurogastroentero. Motil. 21, 952–e76 (2009).
Chassard, C. et al. Functional dysbiosis within the gut microbiota of patients with constipated-irritable bowel syndrome. Aliment. Pharmacol. Ther. 35, 828–838 (2012).
Blachier, F. et al. Luminal sulfide and large intestine mucosa: friend or foe? Amino Acids 39, 335–347 (2010).
Matsunami, M. et al. Luminal hydrogen sulfide plays a pronociceptive role in mouse colon. Gut 58, 751–761 (2009).
Kajander, K. et al. Clinical trial: multispecies probiotic supplementation alleviates the symptoms of irritable bowel syndrome and stabilizes intestinal microbiota. Alimentary Pharmacology & Therapeutics 27, 48–57 (2008).
Saulnier, D. M. et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterol. 141, 1782–1791 (2011).
Marteau, P., Guyonnet, D., Lafaye de Micheaux, P. & Gelu, S. A randomized, double−blind, controlled study and pooled analysis of two identical trials of fermented milk containing probiotic Bifidobacterium lactis CNCM I−2494 in healthy women reporting minor digestive symptoms. Neurogastroentero. Motil. 25, 331–e252 (2013).
Matsuki, T., Watanabe, K., Fujimoto, J., Takada, T. & Tanaka, R. Use of 16S rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 70, 7220–7228 (2004).
Pons, N. et al. Meteor, a platform for quantitative metagenomic profiling of complex ecosystems. Journées Ouvertes en Biologie, Informatique et Mathématiques http://www.jobim2010.fr/sites/default/files/presentations/27Pons.pdf (2010). Date accessed (31/07/2014).
Chervaux, C. et al. Genome sequence of the probiotic strain Bifidobacterium animalis subsp. lactis CNCM I-2494. J. Bacteriol. 193, 5560–5561 (2011).
Letunic, I., Yamada, T., Kanehisa, M. & Bork, P. iPath: interactive exploration of biochemical pathways and networks. Trends Biochem. Sci. 33, 101–103 (2008).
Kobayashi, K. et al. Essential Bacillus subtilis genes. P. Natl. Acad. Sci. USA 100, 4678–4683 (2003).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Res. 17, 377–386 (2007).
Molly, K., Vande, W. M. & Verstraete, W. Development of a 5-step multi-chamber reactor as a simulation of the human intestinal microbial ecosystem. Appl. Microbiol. Biotechnol. 39, 254–258 (1993).
Acknowledgements
We thank the MetaHIT consortium for access to data prior to publication. We thank Aurore Tessier, Christèle Tison, Cyrielle Mariannie, Karine Roy, Stéphanie Cools-Portier for their technical help. This study was funded, in part, by the Metagenopolis grant ANR-11-DPBS-0001 and by Danone Research.
Author information
Authors and Affiliations
Contributions
A.A. collected and prepared the samples. P.V., N.P., S.K., R.B. did the bio-informatic and statistical analyses. P.V., S.K., S.D.E., J.V.H. interpreted the data. P.V., S.K. wrote the manuscript. S.D.E., J.H.V., S.K., D.G. revised the manuscript and P.J.W. critiqued it for clinical content. J.F.M., R.O., D.G., S.K., J.E.T.H.V., L.A.H., P.J.W., S.D.E. conceived, designed and supervised the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Informations
Supplementary Information
Table S2
Supplementary Information
Table S3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Veiga, P., Pons, N., Agrawal, A. et al. Changes of the human gut microbiome induced by a fermented milk product. Sci Rep 4, 6328 (2014). https://doi.org/10.1038/srep06328
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06328
This article is cited by
-
Microbiome composition indicate dysbiosis and lower richness in tumor breast tissues compared to healthy adjacent paired tissue, within the same women
BMC Cancer (2022)
-
Yoghurt consumption is associated with changes in the composition of the human gut microbiome and metabolome
BMC Microbiology (2022)
-
Peptides, Exopolysaccharides, and Short-Chain Fatty Acids from Fermented Milk and Perspectives on Inflammatory Bowel Diseases
Digestive Diseases and Sciences (2022)
-
Symbiosis and the Anthropocene
Symbiosis (2021)
-
Probiotics: a Promising Generation of Heavy Metal Detoxification
Biological Trace Element Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
