
Abstract
The aim of this study was to assess the microbial diversity associated with Lake Nyos, a lake with an unusual chemistry in Cameroon. Water samples were collected during the dry season on March 2013. Bacterial and archaeal communities were profiled using Polymerase Chain Reaction-Denaturing Gradient Gel Electrophoresis (PCR-DGGE) approach of the 16S rRNA gene. The results indicate a stratification of both communities along the water column. Altogether, the physico-chemical data and microbial sequences suggest a close correspondence of the potential microbial functions to the physico-chemical pattern of the lake. We also obtained evidence of a rich microbial diversity likely to include several novel microorganisms of environmental importance in the large unexplored microbial reservoir of Lake Nyos.
Similar content being viewed by others


Introduction
Microorganisms constitute a substantial proportion of the biosphere. Their number is at least two to three orders of magnitude larger than that of all the plant and animal cells combined, constituting about 60% of the earth's biomass1; besides, they are very diverse. This significant representation allows them to stand as controllers of the habitability of the planet through the key roles they play in biogeochemical cycles and food webs2,3. Amongst them are 3 very important groups that evolved from the same ancestor, archaea, bacteria and eukarya. On a genetic basis, they belong to the so called “three-domain system”4. They are at the base of the foodwebs in numerous environments where their several metabolisms mobilise the energy5. Over the past decades, researchers have used these 3 groups of microorganisms to study the aquatic systems. The methods used in microbial ecology progressed from standard culture techniques to modern molecular biology methods. These molecular biological methods are based on the 16S (18S) rRNA genes and revealed that several candidate divisions could not be studied using traditional culture techniques6. This remark makes the molecular techniques in microbial ecology crucial for an intensive characterisation of microbial communities in a given environment. DGGE is one of the numerous fingerprinting approaches that have been designed to study microbial communities, what culture techniques would not allow given that the cultivable fraction represents <1% of the total number of prokaryotic species present in a given sample6. DGGE was used in this study to explore the microbial diversity in Lake Nyos for the first time.
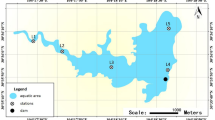
Lake Nyos (Figure 1) is 210 m deep and permanently stratified7,8. Its water column is divided into layers separated by vertical gradients of temperature and dissolved chemical species. The CO2 from deep-magmatic origin reaches the bottom and dissolves into the lake water7,8,9. In 1986, the lake suddenly released a cloud of carbon dioxide into the atmosphere, killing about 1,800 people and 3,000 livestock in nearby towns and villages. This devastating event led to the “Killer Lake” appellation. Since the catastrophe, several studies led to a better understanding of the lake's geological and physico-chemical characteristics. Notably, the CO2 content of the lake increases towards the bottom (the anoxic environment is produced primarily by the absence of dissolved oxygen). Small amounts of endogenic siderite and traces of pyrite have been found in the sediments10. The dissolved chemical species are overwhelmingly dominated by CO2 and HCO3−, followed by Fe2+. Apart from Lake Nyos, two other lakes are known to be stratified and accumulate gas: 1) Lake Monoun in Cameroon and 2) Lake Kivu in the Rwanda-Democratic Republic of Congo border. Among the numerous meromictic lakes worldwide, those three lakes are unique in terms of the amount of gas in the hypolimnion11. In the aftermath of the gas explosion at Lake Nyos, the works of Kling et al.12 and Kusakabe et al.8 showed that the gas started to accumulate again; to avert recurrence of catastrophe, it was suggested to degas the lake. Degassing is done since 2001 and the physico-chemical parameters are continuously monitored12,8.

Geographical location of Lake Nyos (a) and View of the lake in March 2013 (b). OCB: Ocean Continent Boundary; Map: the map was modified from Aka60; Lake's picture: Photo courtesy of Yutaka Yoshida.
Despite the important role that microorganisms play in the geochemical processes, no research has been conducted on the biology of the lake; for that reason, we focused here on the determination of bacterial and archaeal communities and on the understanding of their role in the geochemical processes of the lake.
Results
Physico-chemical characteristics of the samples
Plots of the physico-chemical parameters (Table S1) of the lake in March 2013 are presented in Figure 2 which shows the overwhelming domination of CO2(aq) (a), HCO3− (b) and Fe2+ (c) species. The CO2 concentrations (Figure 2a) increased from 0.2 to 153 mmol kg−1. Similar to the conductivity profile, bicarbonates increased from 2.4 to 38.9 mmol kg−1 with a slight decrease (2 to 1.1 mmol kg−1) from −45 to −80 m (Figure 2b). The Fe2+ concentration was low from 0 to −80 m, then increased considerably from −90 to −210 m (Figure 2c). The other chemical species followed the same trend (increasing but not abruptly as Fe2+) with exception of SO42− and NO3− (Figure 2d), which decreased from the surface to the bottom. The acidity slightly decreased while the temperature slightly increased from about −70 m to the bottom of the lake. Dissolved oxygen (DO) was not detected at the depths where it was measured (−100, −120, −140, −200 and −210 m).

Water depth profiles:
carbone dioxide (CO2) (a); bicarbonates (HCO3−) (bicarbonate values have been multiplied by 100) and conductivity (C25) (b); silica (SiO2), manganese (Mn2+) (manganese values have been multiplied by 10), sodium (Na+) and ferrous iron (Fe2+)(c); sulfate (SO42−) (sulfate values have been multiplied by 10) and nitrate (NO3−) (d); temperature (T) (e) and hydrogen potential (pH) (f).
DGGE profiles of microbial community structures based on 16S rRNA and ecological indexes
The number of detectable bands varied from 6 to 23 per track for bacteria (Figure 3a); for archaea (Figure 4a), fewer bands were observed and varied from 2 to 15 per track. The similarities of all gel tracks were calculated to determine the information content of the banding patterns in terms of the structural diversity of the samples. A cluster analysis of the matrix of similarity values was then performed and visualised in a dendrogram for bacteria (Figure 3b) and archaea (Figure 4b).

Denaturing gradient gel electrophoresis (DGGE) profiles of bacterial communities at different depths in Lake Nyos (a). Dendrogram calculated with the clustering algorithm of Unweighted Pair-Group Method with an Arithmetic Mean (UPGMA) for bacteria in samples from all depths (b). Vertical changes of Shannon-Weaver index of diversity (H’) based on the number and relative intensities of the bands for bacteria identified by DGGE analysis of PCR-amplified 16S rRNA gene (c). NSed: Sediment sample collected at the bottom of the lake. Arabic numerals: Successfully sequenced bands for bacteria. M: Mass ladder standards.

Denaturing gradient gel electrophoresis (DGGE) profiles of archaeal communities at different depths in Lake Nyos (a). Dendrogram calculated with the clustering algorithm of Unweighted Pair-Group Method with an Arithmetic Mean (UPGMA) for archaea in samples from all depths (b). Vertical changes of Shannon-Weaver index of diversity (H’) based on the number and relative intensities of the bands for archaea identified by DGGE analysis of PCR-amplified 16S rRNA gene (c). NSed: Sediment sample collected at the bottom of the lake. Arabic numerals: Successfully sequenced bands for archaea. M: Mass ladder standards.
The bacterial banding pattern was differentiated into four clusters as a function of depth as follows: two upper clusters from 0 to −10 m, −25 to −80 m and two lower clusters from −90 to −160 m and −180 to −210 m. The archaeal banding pattern showed 3 clusters: ranging from −25 to −80 m, −90 to −180 m and −200 to −210 m.
PCA plot performed on the physico-chemical variables per sampling depth (Figure 5) shows the clustering between sampling sites and physico-chemical parameters. The first PC (PC1) displayed a greater variation of the sampling sites and a negative correlation with the physico-chemical parameters, while the second PC (PC2) showed a positive correlation with the parameters. While the majority of the parameters had a very close orientation pattern, SO42− and pH seemed to have a different orientation. The parameters seemed to have more relation with the deepest depths (from −100 to −210 m). MDS plots produced a similar clustering pattern as the dendrograms, showing four clusters for bacteria (Figure 6a) and three for archaea (Figure 6b).

Principal component analysis (PCA) of Lake Nyos showing the depth-related differences and the structuring role of the physico-chemical parameters.
CO2: Carbon Dioxide; HCO3−: Bicarbonates; Fe2+: Ferrous Iron; C25: Conductivity; Mn2+: Manganese; Na+: Sodium; SiO2: Silica; pH: Hydrogen Potential; NO3−: Nitrate and SO42−: Sulfate.

Multidimensional scaling plots produced to compare the sampling depths for bacteria (a) and archaea (b).
NSed: Sediment sample collected at the bottom of the lake.
Bacterial and archaeal community structure and distribution
A total of 46 bands were successfully sequenced for bacteria and 15 bands for archaea. Bacteria showed a higher diversity (Figure 3c) than archaea (Figure 4c), with a DGGE profile that was far more complex. The successfully sequenced bands are indicated with arabic numerals on the DGGE gels (Figures 3a and 4a). The obtained sequences were aligned to the GenBank sequences of the 16S rRNA gene for bacteria and archaea database; the nearest relatives were identified for bacterial (Table 1) and archaeal sequences (Table 2). All the sequences that were ≥97% similar to the same organism were grouped under the same genotype and named as Nyos bacteria (Table 1) or Nyos archaea (Table 2) genotypes. Both bacteria and archaea domains exhibited distinct communities at different depths of the lake's water column and their dendrograms indicated a stratified pattern along the water column (Figures 3b and 4b) with the largest difference occuring around 80–90 m depth. The bulk of the matches of bacterial sequences belonged to the phyla Firmicutes and Actinobacteria that accounted each for about 28.3%. They dominated from 0 to −55 m, while fewer representatives were found at other depths. However, when downscaling, some differences arose to finer taxonomic resolution. The matches of the detected sequences belonged to the phyla Proteobacteria (21.7%), Bacteroidetes (8.7%), Caldiserica (4.4%) and 2.2% for each of the phyla Ignavibacteriae, Nitrospirae, Tenericutes and Fusobacteria. For the archaeal sequences, the closest relatives belonged to the Thaumacheota (53.3%), Euryarchaeota (33.3%) and Crenarcheota (13.3%).
Relationship between bacterial and archaeal communities with the various depths
The vertical profiles of the physicochemical parameters of the lake showed a stratification with a considerable increase of concentrations starting from about −80 m for CO2, HCO3−, Fe2+ and C25. Mn2+, Na+ and SiO2 concentrations slightly decreased around −60 m and increased again around −100 m. NO3− and SO42− concentrations rather kept a reducing trend along the column towards the bottom and almost stabilised at −80 m. Na+ concentration was very low in the upper part of the lake and started increasing gradually around −80 m to reach its maximum concentrations towards the bottom. These chemical features could map with the identified bacterial and archaeal sequences, suggesting a close relationship between the potential functions of the bacterial/archaeal communities and the physico-chemical characteristics of the various layers of the lake. Even though the archaeal community was less diverse, its stratification pattern was clearly displayed.
The bacterial bands obtained from 0 to −35 m were highly similar to the aerobic Exiguobacterium sp. and Arthrobacter sp.; sequences close to Micropruirina glycogenica, a facultative anaerobic bacterium and capable to reduce nitrate as mentioned by Shintani et al.13 were also present. Organisms close to the Acidimicrobium ferrooxidans, capable of ferrous iron oxidation and carbon dioxide fixation14 were found at −45, −120, −160 and −180 m. Moreover, a sequence close to Ferrithrix thermotolerans strain Y005 was detected at −45 m; the latter is able to reduce and oxidise iron; furthermore, it is capable of oxidative dissolution of pyrite15. Melioribacter roseus strain P3M may also be able to use ferric iron as electron acceptor as mentioned by Kadnikov et al.16 and corresponded to the closest match of one of the bands from the −120 m sample. A sequence related to Methylocystis echinoides strain IMET 10491 was found at −70m; the latter is an anaerobic organism capable of methane-oxidation17. Sequences matching with the thiosulfate oxidising and carbon dioxide fixing bacteria Thiobacillus thiophilus strain D24TN18 were found at −80 m. Sequences of the genotype 26 that appeared among the bands generated from the sample of −180 m could also play a part in the thiosulfate metabolism as they matched with Caldisericum exile strain AZM16c01T isolated from a hot spring in Japan and reported as thiosulfate-reducing bacterium19. Sequences close to the bacteria (Desulfovibrio vulgaris strain DP4 (−160 m), Desulfotomaculum australicum strain AB33 (−100), Desulfovibrio alaskensis strain G20 (−160 and −180 m)) that contribute to sulfate metabolism as reported by Zane et al.20, Love et al.21 and Hauser et al.22, respectively and the bacteria (Candidatus Nitrospira defluvii (−180 m), Bacillus alkalinitrilicus strain ANL-iso4 (−140 m), Melioribacter roseus strain P3M (−120 m)) involved in the nitrogen metabolism as mentioned by Lücker et al.23, Sorokin et al.24 and Podosokorskaya et al.25, respectively were detected. One of the bands sequenced at −140 m was the close relative of Clostridium cellulolyticum H10, reported to produce H226. Another putative hydrogen producer detected in this study could be NyosB46 genotype 33 from the sediment sample since its sequence matched with Psychrilyobacter atlanticus strain HAW-EB21, a hydrogen (H2) producer27.
Archaeal sequences dominating from −10 to −70 m were closely related to the ammonia oxidising archaeon Candidatus Nitrosopumilus koreensis strain AR1. They inhabit various environments among which marine water, fresh water and hot springs and play important roles in the global nitrogen and carbon cycles28,29,30. The sequenced band from −80 m revealed a similarity of 87% to Methanolobus psychrophilus R15, a methanogen31. The sequences of bands from −120 and −180 m matched with Natronolimnobius baerhuensis IHC-005, a species isolated from soda lakes capable to reduce nitrate to nitrite and thiosulfate or sulfur to sulfide32. Bands from the −210 m (water and sediment sample) resulted in sequences matching with the Euryarchaeota Vulcanisaeta distributa DSM 14429 strain IC-017 and Salinarchaeum sp. Harcht-Bsk1.
Discussion
Bacterial and archaeal communities detected in this study showed a stratified distribution in the water column agreeing with previous studies using physico-chemical approaches. Several genera belonging to diverse functional groups were detected. This diversity of microbial communities likely reflects the uniqueness of Lake Nyos. However, it is worth mentioning that artifacts may be introduced in the microbial assemblages during DNA extraction, PCR or DGGE and therefore alter the natural diversity of microbial communities33. Consequently, the microbial community composition as detected by DGGE in this study may mainly represent the most abundant phylotypes. If functionally active, they could considerably impact the sulfur, hydrogen, nitrogen, methane and carbon cycles taking place in the lake.
Altogether, the dendrograms showed clusters of the water stratification following the physico-chemical pattern indicated by Kusakabe et al.8; this clustering is also well illustrated by the MDS analysis with nearly similar clusters. Microorganisms require different metabolic pathways to survive and differences in concentrations of the chemical species may, to a large extent, explain differences in the bacterial and archaeal communities between the water layers. This remark is supported by the disposition of the sampling depths on relatively well defined loci on the PCA plot. The shallowest and deepest sampling sites are well separated, indicating the structuring role of the physicochemical parameters that have likely influenced the microbial community distribution.
The taxonomic structure of both bacterial and archaeal communities assemblage was nearly similar, with clear phylogenetic distribution influenced by the oxic or anoxic state of the sampling depths. Sequences found from 0 to −45 m were exclusively closer to aerobic species; from −55 to −180 m, they matched with both aerobic and anaerobic species and under −180 m, they were all close to anaerobes. The deep layers of Lake Nyos are known to be anoxic8, as demonstrated by our measurements. The presence of such potential aerobic species might suggest that minute amounts of oxygen may be found up to deep water layers in the lake.
Apart from the CO2 concentration, many other environmental variables were linked to the vertical stratification of the studied communities as typically seen in meromictic lakes. Sequences close to the phylogenetic groups Actinobacteria, Firmicutes, Nitrospirae and Proteobacteria were detected as in Lake Tanganyika34; archaeal sequences related to Thaumarchaeota were found in the upper part of the lake, as in the case of other meromictic lakes such as Lake Kivu35,36 and Lake Ace37 and marine environments such as the northern South China Sea38.
Genetic fingerprinting techniques are key tools for studies of microbial communities, but do not provide direct evidence of a community's functional capabilities. Assurances on functionality of genes encoded in a microorganism's genome could only be reached through culture techniques or studies based on RNA analyses. However, functionality of the genome of a detected organism may be predicted on the basis of functions encoded in closely related genomes39. The microbial sequences retrieved from Lake Nyos water column were related to a variety of different functional groups of which a speculative correlation with the lake water's chemistry is discussed in the following lines.
Chemical reactions involving iron species in the lake could be governed by the combined contribution of sequences related to A. ferrooxidans strain DSM 10331 (at −45, −120, −160 and −180 m) and F. thermotolerans strain Y005 (at −55 m). The gradual increase of Fe2+ concentration towards the bottom of the lake as observed on Figure 2c could result from the concomitant actions of these iron related species. The vertical plot of Fe2+ concentration along the water column illustrates that iron is one of the most important chemical species in Lake Nyos. In fact, the iron cycle principally features the interchange of Fe2+ to Fe3+ and vice versa. With respect to the putative iron metabolising bacteria that were detected in the water column, both reactions would be expected to take place. But the increasing concentration of Fe2+ with depth suggests that the most favorable reaction would be the reduction of Fe3+ to Fe2+, which may contribute to the siderite formation at the bottom of the lake observed by Bernard and Symonds10. This hypothesis is supported by the study of Ellwood et al.40 stating that the formation of siderite from Fe2+ produced during bacterial dissimilatory iron reduction is plausible in anoxic sediments. In fact, dissimilatory reduction occurs when ferric iron serves as a terminal electron acceptor during anaerobic respiration41. Under such conditions, pyrite mentioned by Bernard and Symonds10 may likely form as well by using the sulfur species produced in the sulfur cycle.
Apart from the ability of these two phylogenetic groups to transform iron species, they have been reported to fix CO242 as also may do the sequences related to T. thiophilus strain D24TN15,18 at 80 m. If active in the Lake Nyos water column, such CO2 fixing bacteria would likely contribute to the decrease of the CO2 concentrations, even though at a micro level.
In the anoxic layers of Lake Nyos, several genotypes with putative activity in the sulfur cycle were detected. Such microorganisms have been reported in Lake Cadagno43 and Lake Ace37. Sulfate ion is a primary factor in the distribution of microbial activities in anoxic sediments. When sulfate diffuses into anoxic habitats, it provides an opportunity for different groups of microorganisms to carry out dissimilatory sulfate reduction. This results in sulfide accumulation. Sulfide can serve as an electron source for anoxygenic chemolithoautotrophs such as Thiobacillus. SO42− is a common source of energy for anaerobic sulfobacteria. These biological processes produce reduced sulfur species as well as elemental sulfur44,45. In our study, the genotypes that could putatively act on sulfur species are the archaeal sequences detected at −120 and −180m and the bacterial sequences detected at −80, −100, −160 m as previously mentioned. However, sulfur is depleted along the water column and this could limit the dissimilatory capacity of the sulfate related microorganisms36,46. If functional in the lake, they may degrade a wide variety of organic compounds heterotrophically or grow autotrophically, fixing inorganic CO2 into central metabolic intermediates, given that they are metabolically versatile47.
Two sequences close to species related to the methane metabolism were found at −70 and −80 m. The presence of methane in the lake have been mentioned and thought to be biogenic12,48,49. The NyosA9 genotype 2 could be a putative producer of the methane present in the lake, although its origin has not yet been determined. A non-sequenced band appears at the same position on the DGGE gel (archaea) for the sample collected at −90 m. We also specify that all the archaeal bands sequenced from −120 to −210 m matched with the unclassified anaerobic methanogenic archaeon ET1-10 in the nucleotide collection database. The similarity was quite high (86–99%) with respect to the similarity obtained in the 16S rRNA gene database for bacteria and archaea. This suggests that the organisms could correspond to a methanogenic archaeon, rather than Vulcanisaeta distributa. The detection of such putative methanogenic organism appears to answer the question of methane source in Lake Nyos. Safety concerns have been raised by Issa et al.49 regarding the increasing concentration of CH4 in the lake, since it has a low solubility. If the methanotrophic related bacteria detected are functionally active, their presence in the lake could be reassuring because they would feed on the available methane, contributing hereby to its reduction. Consequently, the methane present in the lake would be affected by methanogenesis and methanotrophy reactions performed by the detected microorganisms, subject to their functionality and abundance. The microbial methane production could also be influenced by the sulfur cycle. For instance, methanogenesis is responsible of the majority of terminal metabolism under anoxic conditions in freshwaters. However, methanogenesis and sulfate respiration compete for the same substrates. In such conditions, sulfate respiration likely dominates on the methanogenesis50,51. When enough sulfate-reducing bacteria are present, they maintain the concentrations of hydrogen and acetate at levels too low for methanogens to grow52. Part of the methane could also be oxidised into HCO3− and HS by the putative sulfate-reducing bacteria53 detected in the lake. Taking into account these hypotheses and the presence of methanotrophic bacteria, we could expect that the methane concentration is kept low. A specific study using primers to screen methane metabolising archaea and bacteria, as well as quantitative analyses of methane and sulfur bacteria, would be needed to test these hypotheses.
Na+ concentrations increased towards the bottom of the lake, with the highest concentrations in the deepest layers (−207 to −210 m); the sequence of one strong band of the bottom sample matched with Salinarchaeum sp. strain HArcht-Bsk1 (91%). Together with the sequences from −120 and −180 m, matching with N. baerhuensis IHC-005 (88–92%), they constitute putative halobacteriaceae species that could exist in the deepest layers of Lake Nyos. Salinarchaeum sp. strain HArcht-Bsk1 was isolated from Lake Baskunchak (Russia), with a hypersaline chloride-sulfate environment54; and N. baerhuensis IHC-005 from soda lakes in Inner Mongolia32.
Several other functions such as the phosphate metabolism could also take place in the lake water column as related organisms (NyosB7 genotype 3_1 and NyosB12 genotype 3_2) close to M. glycogenica13 were detected.
We used molecular techniques to study the microbial diversity in a meromictic lake water column and sediments. The archaeal and bacterial communities were revealed for the first time and showed a stratified pattern that mapped with the limnological conditions of the lake. The potential interactions of these communities with the chemical species of the lake's water column have been discussed. This study is a starting point of a broad descriptive microbiology in Cameroon's lakes and other water ecosystems. Many putative biotechnologically interesting microorganisms, with genes sufficiently different (from their closest matches in the GenBank) to be novel species candidates have been detected. This pioneer inventory will serve as a guide for further studies. The sequences corresponding to potential CO2, CH4, or Fe metabolising bacteria/archaea could be used in the bio-monitoring of these chemically important species of the lake. Biotechnological use in the removal of CO2 could be one of the future interests.
However, this study may have been affected by technical limitations, such as biases that could be introduced by PCR or DGGE. Therefore, we obviously have got the bacterial community as determined by DGGE, but this does not insure the disclosure of the whole bacterial and archaeal communities. High throughput studies using sharper methods including next generation sequencing would provide more complete information on the microbial communities; furthermore, a three-pronged analytical approach based on the 16S rRNA studies, genes quantification and specific detection is recommended to clarify the functionality and abundance of the species and to characterise the organisms responsible of key biogeochemical functions taking place in the lake.
Methods
Site description
Lake Nyos is a crater lake located in the Northwest of Cameroon. It lies within the Oku volcanic field, along the Cameroon Volcanic Line (CVL) which runs from the Atlantic Ocean to the interior of Cameroon (Figure 1). The volcanism of the CVL is mostly basaltic and dated about 4,000 years55.
Sample collection and processing
The samples were collected at the end of the dry season (March 2013) from the centre of the lake (N06°26′23.0″ and E10°18′02.3″) every 10, 15 or 20 m depth along the water column using a 1.6 l Niskin bottle. Each sample was transferred to a 1 l sterile polypropylene bottle and immediately kept in an ice-cooled box. Subsequently, they were vacuum filtered with a 0.22 µm membrane filter, then stored frozen until DNA extraction. Simultaneously with the water sampling, a profile of the water column was done using a conductivity-temperature-depth (CTD, Ocean Seven 316, Idronaut, Italy) profiler fitted with sensors to measure the conductivity, temperature, pressure (which is later converted to depth), pH and redox potential. DO concentration was measured at the depths of 100, 120, 140, 180, 200 and 210 m using the modified Winkler titration method56.
DNA extraction
Total DNA was extracted from the membrane filters using a soil sample DNA extraction kit (UltraClean® Soil DNA Isolation Kit; Mo Bio Laboratories, CA, USA) and following the manufacturer's instructions. The genomic DNA was then quantified with a spectrophotometer (Nanodrop 1000 Spectrophotometer; Thermo Fisher Scientific K.K., Kanagawa, Japan) and stored at −20°C for later use.
Small Subunit rRNA gene amplification, DGGE analysis and sequencing
PCR amplification was performed for the domains bacteria and archaea in a 40 µl volume containing approximately 5 ng template DNA, 200 mmol l−1 of each of the deoxynucleoside triphosphates, 250 µmol l−1 of each of the primers, 1x Ex-Taq buffer and 0.5 U of DNA Taq polymerase (Takara Bio, Inc., Shiga, Japan). More information on the primers used in this study is given in supplementary Table S2. To analyse the bacterial diversity, the universal primers GC-341f and 518r57 were used under the following thermal cycles: the initial denaturation at 94°C for 3 min; 20 touchdown cycles of denaturation (at 98°C for 10 sec), annealing (at 65°C for 30 sec, decreasing 0.5°C each cycle) and extension (at 72°C for 30 sec); 15 standard cycles of denaturation (at 98°C for 10 sec), annealing (at 55°C for 30 sec and extension (at 72°C for 30 sec) and a final extension at 72°C for 3 min. For the archaeal 16S rRNA genes, a nested PCR using the primers sets 20f and 958r57 in the first round of PCR and the inner primers GC-340f and 519r57 in the second round with the following for both: the initial denaturation at 94°C for 3 min; 10 touchdown cycles of denaturation (at 94°C for 1 min), annealing (at 53°C for 1 min, decreasing 1°C each cycle) and extension (at 72°C for 3 min). Successful DNA amplification was verified by electrophoresis of 2 µl of PCR products in 2% agarose gel in TAE buffer (40 mM Tris-acetate, 1 mM EDTA, pH 8.0) with a DNA Mass Ladder Standard (Nippon Gene, Tokyo, Japan).
The remaining 38 µl of the amplified 16S rRNA gene fragments were analysed by DGGE with a DcodeTM Population Base System (Bio-Rad, Laboratories, CA, USA). PCR products were separated with an 8% acrylamide gel on a linear gradient of the denaturants urea and formamide increasing from 25% at the top of the gel to 60% at the bottom and ran as described by Muyzer et al.58 with little modifications. Samples were analysed simultaneously with a marker at both extreme wells. The gel was stained with ethidium bromide and documented using a UV-transilluminator (MultiDoc-ItTM Digital Imaging System; UVP, CA, USA) and a C-5060 Wide Zoom imaging systems (Olympus, Tokyo, Japan).
DGGE pattern analysis, phylogenetic analysis of excised bands and statistical analysis
The positions and relative signal intensities of detected bands in each gel track were determined with FP Quest software (Bio-Rad, Laboratories, CA, USA). Relative signal intensities were calculated from the peak area of the densitometric curves. Clustering analysis of the DGGE banding pattern was performed with the unweighted pair-group method using arithmetic averages (UPGMA) and the FP Quest software package was used for dendrogram construction. The Shannon-Weaver index (H’) was used as an estimate of microbial diversity. H’ was calculated using the following equation59:

Where, pi represents the relative signal intensities of bands in a track and R, the Richness. All the detected bands were used for the calculation of diversity indices.
We applied principal component analysis (PCA) and the MDS algorithm to explore variation in the data with the PRIMER software (version 2, PRIMER-E Ltd, Plymouth, UK). For the PCA, the data matrix used the physico-chemical parameters as the variables at each depth. For the MDS, depths were used as the variables, the band scores as the values within each variable and the correlation coefficient to calculate the similarity matrix. The first two components (Supplementary Table S3) were used to interpret the results.
The strong bands were stabbed with a sterile pipette tip; each stab was placed into 1.5 ml eppendorf tubes with 100 µl of sterile water and stored overnight at 4°C. After an amplification check, the bands were purified on Qiaquick columns (Qiagen, Tokyo, Japan) and a BigDye PCR (Applied Biosystems, CA, USA) performed. Then they were sequenced on an ABI-Prism sequencer (Applied Biosystems) using the reverse primer 518r for bacterial sequences and 519r for archaeal sequences. The obtained sequences were manually edited in the ATGC software. Subsequently, they were submitted to the GenBank database of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov) for alignment employing the Basic Local Alignment Search Tool (BLASTN) algorithm of nucleotide in the 16S rRNA sequences (Bacteria and Archaea) database to determine their phylogenetic affiliations. The nearest neighbours of the submitted sequences, with the lowest E-value were considered as closest taxonomic affiliates.
Nucleotide sequence accession numbers
The sequences generated in this study were deposited at the GenBank/EMBL/DDBJ databases under the accession numbers AB907636 to AB907681 for bacteria; AB907765 to AB907774 and AB917141 to AB917145 for archaea.
References
Singh, B. K., Campbell, C. D., Sorenson, S. J. & Zhou, J. Soil genomics. Nature Rev. Microbiol. 7, 756 (2009).
Zehr, J. P. Microbes in Earth's aqueous environments. Front. Microbiol. 1, 4 (2010).
Hoegh-Guldberg, O. & Bruno, J. F. The impact of climate change on the world's marine ecosystems. Science 328, 1523–1528 (2010).
Woese, C., Kandler, O. & Wheelis, M. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya. Proc. Natl. Acad. Sci. USA 87, 45769 (1990).
Jansson, M., Persson, L., De Roos, A. M., Jones, R. I. & Tranvik, L. J. Terrestrial carbon and intraspecific size-variation shape lake ecosystems. Trends. Ecol. Evol. 22, 316–322 (2007).
Rastogi, G. & Sani, R. K. Molecular techniques to assess microbial community structure, function and dynamics in the environment. in Microbes and Microbial Technology: Agricultural and Environmental Applications (Ahmad, I., Ahmad, F., Pichtel, J. (ed.)) 29–57 (Springer, New York, 2011).
Kling, G. W., Tuttle, M. L. & Evans, W. C. The evolution of thermal structure and water chemistry in Lake Nyos. J. Volcanol. Geotherm. Res. 39, 151–165 (1989).
Kusakabe, M. et al. Evolution of CO2 in Lakes Monoun and Nyos, Cameroon, before and during controlled degassing. Geochem. J. 42, 93–118 (2008).
Nagao, K., Kusakabe, M., Yoshida, Y. & Tanyileke, G. Noble gases in Lakes Nyos and Monoun, Cameroon. Geochem. J. 44, 519–543 (2010).
Bernard, A. & Symonds, R. B. The significance of siderite in the sediments from Lake Nyos, Cameroon. J. Volcanol. Geoth. Res. 39, 187–194 (1989).
Tassi, F. et al. Water and gas chemistry at Lake Kivu (DRC): Geochemical evidence of vertical and horizontal heterogeneities in a multibasin structure. Geochem. Geophys. Geosyst. 10, 10 (2009).
Kling, G. W. et al. Degassing Lakes Nyos and Monoun: Defusing certain disaster. PNAS 102, 14185–14190 (2005).
Shintani, T. et al. Micropruina glycogenica gen. nov., sp. nov., a new Gram-positive glycogen-accumulating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 50, 201–207 (2000).
Clark, D. A. & Norris, P. R. Acidimicrobium ferrooxidans gen. nov., sp. nov.: mixed-culture ferrous iron oxidation with Sulfobacillus species. Microbiol. 142, 785–790 (1996).
Johnson, D. B., Bacelar-Nicolau, P., Okibe, N., Thomas, A. & Hallberg, K. B. Ferrimicrobium acidiphilumgen nov., sp. Nov. and Ferrithrix thermotolerans gen. nov., sp. Nov.: heterotrophic, iron-oxidizing, extremely acidophilic actinobacteria. Int. J. Syst. Evol. Microbiol. 59, 1082–1089 (2009).
Kadnikov, V. V. et al. Genomic analysis of Melioribacter roseus, facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi group. PloS ONE 8, e53047 (2013).
Heyer, J., Galchenko, V. F. & Dunfield, P. F. Molecular phylogeny of type II methane-oxidizing bacteria isolated from various environments. Microbiol. 148, 2831–2846 (2002).
Kellermann, C. & Griebler, C. Thiobacillus thiophilus sp. nov., a chemolithoautotrophic, thiosulfate-oxidizing bacterium isolated from contaminated aquifer sediments. Int. J. Syst. Evol. Microbiol. 59, 583–588 (2009).
Mori, K., Yamaguchi, K., Sakiyama, Y., Urabe, T. & Suzuki, K. Caldisericum exile gen. nov., sp. nov., an anaerobic, thermophilic, filamentous bacterium of a novel bacterial phylum, Caldiserica phyl. nov., originally called the candidate phylum OP5 and description of Caldisericaceae fam. nov., Caldisericales ord. nov. and Caldisericia classis nov. Int. J. Syst. Evol. Microbiol. 59, 2894–8 (2009).
Zane, G. M., Yen, H. C. & Wall, J. D. Effect of the deletion of qmoABC and the promoter-distal gene encoding a hypothetical protein on sulfate reduction in Desulfovibrio vulgaris Hildenborough. Appl. Environ. Microbiol. 76, 5500–5509 (2010).
Love, A. C., Patel, B. K., Nichols, P. D. & Stackebrandt, E. Desulfotomaculum australicum, sp. nov., a thermophilic sulfate-reducing bacterium isolated from the Great Artesian Basin in Australia. Syst. Appl. Microbiol. 16, 244–251 (1993).
Hauser, L. J. et al. Complete genome sequence and updated annotation of Desulfovibrio alaskensis G20. J. Bacteriol. 193, 4268–4269 (2011).
Lücker, S. et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. PNAS 107, 13479–13484 (2010).
Sorokin, D. Y., van Pelt, S. & Tourova, T. P. Utilization of aliphatic nitriles under haloalkaline conditions by Bacillus alkalinitrilicus sp. nov. isolated from soda solonchak soil. FEMS Microbiol. Lett. 288, 235–240 (2008).
Podosokorskaya, O. A. et al. Characterization of Melioribacter roseus gen. nov., sp. nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria and a proposal of a novel bacterial phylum Ignavibacteriae. Environ. Microbiol. 15, 1759–1771 (2013).
Guedon, E., Payot, S., Desvaux, M. & Petitdemange, H. Relationships between cellobiose catabolism, enzyme levels and metabolic intermediates in Clostridium cellulolyticum grown in a synthetic medium. Biotechnol. Bioeng. 67, 327–335 (2000).
Zhao, J. S., Manno, D. & Hawari, J. Psychrilyobacter atlanticus gen. nov., sp. nov., a marine member of the phylum Fusobacteria that produces H2 and degrades nitramine explosives under low temperature conditions. Int. J. Syst. Evol. Microbiol. 59, 491–497 (2009).
Leininger, S. et al. Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442, 806–809 (2006).
Park, S. J., Park, B. J. & Rhee, S. K. Comparative analysis of archaeal 16S rRNA and amoA genes to estimate the abundance and diversity of ammonia-oxidizing archaea in marine sediments. Extremophiles 12, 605–615 (2008).
Park, S. J. et al. Draft genome sequence of an ammonia-oxidizing archaeon, “Candidatus Nitrosopumilus koreensis” AR1, from Marine Sediment. J. Bacteriol. 194, 6940–6941 (2012).
Zhang, G., Jiang, N., Liu, X. & Dong, X. Methanogenesis from Methanol at Low Temperatures by a Novel Psychrophilic Methanogen, “Methanolobus psychrophilus” sp. nov., Prevalent in Zoige Wetland of the Tibetan Plateau. Appl. Environ. Microbiol. 74, 6114–6120 (2008).
Itoh, T., Yamaguchi,, T., Zhou, P. & Takashina, T. Natronolimnobius baerhuensis gen. nov., sp. nov and Natronolimnobius innermongolicus sp. nov., novel haloalkaliphilic archaea isolated from soda lakes in Inner Mongolia, China. Extremophiles 9, 111–116 (2005).
Casamayor, E. O. et al. Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ. Microbiol. 4, 338–348 (2002).
De Wever, A. et al. Bacterial Community Composition in Lake Tanganyika: Vertical and Horizontal Heterogeneity. Appl. Environ. Microbiol. 71, 5029–5037 (2005).
Llirós, M. et al. Vertical distribution of ammonia-oxidizing crenarchaeota and methanogens in the epipelagic waters of Lake Kivu (Rwanda-Democratic Republic of the Congo). Appl. Environ. Microbiol. 76, 6853–6863 (2010).
Llirós, M. et al. Microbial Ecology of Lake Kivu, in Lake Kivu: Limnology and biogeochemistry of a tropical great lake. (Descy, J. P.,Darchambeau, F., Schmid, M. (ed.)) 85–105 (Springer, Netherlands, 2012).
Comeau, A. M., Harding, T., Galand, P. E., Vincent, W. F. & Lovejoy, C. Vertical distribution of microbial communities in a perennially stratified Arctic lake with saline, anoxic bottom waters. Sci. Rep. 2, 604; 10.1038/srep00604 (2012).
Dang, H. et al. Thaumarchaeotal signature gene distribution in sediments of the northern South China Sea: an indicator of the metabolic intersection of the marine carbon, nitrogen and phosphorus cycles? Appl. Environ. Microbiol. 79, 2137–47 (2013).
Langille, M. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013).
Ellwood, B. B. Chrzanowski, H. T., Hrouda, F., Long, G. J. & Buhl, M. L. Siderite formation in anoxic deep-sea sediments: A synergetic bacteria controlled process with important implications in paleomagnetism. Geol. 16, 980–982 (1988).
Liermann, L. J., Hausrath, E. M., Anbar, A. D. & Brantley, S. L. Assimilatory and dissimilatory processes of microorganisms affecting metals in the environment. J. Anal. Atom. Spectrom. 22, 867–877 (2007).
Esparza, M., Cárdenas, J. P., Bowien, B., Jedlicki, E. & Holmes, D. S. Genes and pathways for CO2 fixation in the obligate, chemolithoautotrophic acidophile, Acidithiobacillus ferrooxidans, Carbon fixation in A. ferrooxidans. BMC Microbiol. 10, 229 (2010).
Tonolla, M., Peduzzi, S., Demarta, A., Peduzzi, R. & Hahn, D. Phototropic sulfur and sulfate-reducing bacteria in the chemocline of meromictic Lake Cadagno, Switzerland. J. Limnol. 63, 161–170 (2004).
Kallistova, A. Y. et al. Sulfate reduction and methanogenesis in the Shira and Shunet meromictic lakes (Khakasia, Russia). Microbiology 75, 720–726 (2006).
Tassi, F. et al. Morphological and geochemical features of crater lakes in Costa Rica: an overview. J. Limnol. 68, 193–205 (2009).
Rankin, L. M., Gibson, J. A. E., Franzmann, P. D. & Burton, H. R. The chemical stratification and microbial communities of Ace Lake, Antarctica: A review of the characteristics of a marine-derived meromictic lake. Polarforschung 66, 33–52 (1999).
Londry, K. L. & Des Marais, D. J. Stable carbon isotope fractionation by sulfate-reducing bacteria. Appl. Environ. Microbiol. 69, 2942–2949 (2003).
Evans, W. C. et al. Six years of change at Lake Nyos, Cameroon, yield clues to the past and cautions for the future. Geochem. J. 28, 139–162 (1994).
Issa et al. Contribution of methane to total gas pressure in deep waters at Lakes Nyos and Monoun (Cameroon, West Africa). Geochem. J. 47, 349–62 (2013).
Winfrey, M. R. & Zeikus, J. G. Effect of sulfate on carbon and electron flow during microbial methanogenesis in freshwater sediments. Appl. Environ. Microbiol. 33, 275–281 (1977).
Capone, D. G. & Kiene, R. P. Comparison of microbial dynamics in marine and freshwater sediments: Contrasts in anaerobic carbon catabolism. Limnol. Oceanogr. 33, 725–749 (1988).
Lovley, D. R. & Klug, M. J. Model for the distribution of sulfate reduction and methanogenesis in freshwater sediments. Geochim. Cosmochim. 50, 11–18 (1986).
Barton, L. L. & Fauque, G. D. Biochemistry, Physiology and Biotechnology of Sulfate-Reducing Bacteria. Adv. Appl. Microbiol. 68, 41–98 (2009).
Dominova, I. N., Sorokin, D. Y., Kublanov, I. V., Patrushev, M. V., Toshchakov, S. V. Complete genome sequence of Salinarchaeum sp. strain HArcht-Bsk1T, isolated from hypersaline Lake Baskunchak, Russia. Genome Announc. 1, 505; 10.1128/genomeA00505 (2013).
Aka, F. T. et al. U-series dating of Lake Nyos maar basalts, Cameroon (West Africa): Implications for potential hazards on the Lake Nyos dam. J. Volcanol. Geoth. Res. 176, 212–224 (2008).
Carpenter, J. H. The Chesapeake Bay Institute technique for the Winkler dissolved oxygen method. Limnol. and Oceanogr. 10, 141–143 (1965).
Nam, Y. D. et al. Bacterial, archaeal and eukaryal diversity in intestines of Korean people. J. Microbiol. 46, 491–501 (2008).
Muyzer, G., de Waal, E. C. & Uitterlinden, A. G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59, 695–700 (1993).
Andreoni, V. et al. Bacterial communities and enzyme activities of PAHs polluted soils. Chemosphere 57, 401–12 (2004).
Aka, F. T. Noble gas systematics and K-Ar chronology: Implications for the Geochemical and Geotectonic Evolution of the Cameroon Volcanic Line, West Africa. Doctoral Thesis. University of Okayama, Japan (2000).
Acknowledgements
This research was funded by the SATREPS-NyMo project. We sincerely thank all the members of the project. An additional support was given by Nakamura's laboratory. We are indebted to Prof Kuate Jules Roger and Dr Tume Christopher of the laboratory of Biochemistry of the University of Dschang (Cameroon) for their support.
Author information
Authors and Affiliations
Contributions
This work was conceived by R.E.T., S.N. and A.U. The sampling was done by R.E.T., W.F., M.K., G.T., B.T. and V.H. M.K. and T.O. generated the physico-chemical data. Molecular analyses were done by R.E.T., A.S., A.N. and D.T. The manuscript was written and approved by all the authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Tables and Legend
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Tiodjio, R., Sakatoku, A., Nakamura, A. et al. Bacterial and archaeal communities in Lake Nyos (Cameroon, Central Africa). Sci Rep 4, 6151 (2014). https://doi.org/10.1038/srep06151
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06151
This article is cited by
-
Spatial variation of airborne bacterial heterogeneity and potential opportunistic human pathogens: a comparative study of sites in Korea and Japan
Aerobiologia (2024)
-
Bacterial and archaeal communities within the alkaline soda Langaco Lake in the Qinghai-Tibet Plateau
Annals of Microbiology (2022)
-
Discovery and biogeochemistry of asphalt seeps in the North São Paulo Plateau, Brazilian Margin
Scientific Reports (2018)
-
Taxonomical Resolution and Distribution of Bacterioplankton Along the Vertical Gradient Reveals Pronounced Spatiotemporal Patterns in Contrasted Temperate Freshwater Lakes
Microbial Ecology (2018)
-
Distinct distribution patterns of prokaryotes between sediment and water in the Yellow River estuary
Applied Microbiology and Biotechnology (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
