
Abstract
Peptide rational design was used here to guide the creation of two novel short β-lactamase inhibitors, here named dBLIP-1 and -2, with length of five amino acid residues. Molecular modeling associated with peptide synthesis improved bactericidal efficacy in addition to amoxicillin, ampicillin and cefotaxime. Docked structures were consistent with calorimetric analyses against bacterial β-lactamases. These two compounds were further tested in mice. Whereas commercial antibiotics alone failed to cure mice infected with Staphylococcus aureus and Escherichia coli expressing β-lactamases, infection was cleared when treated with antibiotics in combination with dBLIPs, clearly suggesting that peptides were able to neutralize bacterial resistance. Moreover, immunological assays were also performed showing that dBLIPs were unable to modify mammalian immune response in both models, reducing the risks of collateral effects. In summary, the unusual peptides here described provide leads to overcome β-lactamase-based resistance, a remarkable clinical challenge.
Similar content being viewed by others

Introduction
Antimicrobial resistance is not a new problem, but the number of resistant organisms and lethal outbreaks is unprecedented1,2,3. Infectious agents that were once supposed to be controlled by antibiotics are returning in new forms resistant to conventional therapies, clearly making efficient and stable control of microorganisms difficult4,5. Among the antibacterial therapies, the most often used antibiotics commonly consist of lactam, including penicillins and cephalosporins6. Nevertheless, due to widespread use of lactam antimicrobials and also to genetic and biochemical factors, bacterial resistance represents a serious threat to the continuing use of antibiotic treatment7. The most conventional bacterial mechanism of resistance to lactam antibiotics is synthesis of β-lactamases, which are able to cleave the amide bond in the target β-lactam ring, rendering these antibiotics ineffective8. In this context, one logical strategy has been to pursue β-lactamase inhibitors as additives of lactams, to prevent or reduce cleavage of the β-lactam ring9,10.
Results and Discussion
Rational design for constructing synthetic peptides was first based on the pocket volume and distances between the amino acid residues that compose the β-lactamase catalytic triad (KSG) and amino acid residues adjacent to the catalytic triad. Evaluation by docking studies of inhibitor-enzymes was carried out by designing short and flexible synthetic peptide inhibitors that probably interact with the amino acid residues near the β-lactamase catalytic triad. Thus, two peptides named dBLIP-1 and -2 (designated β-lactamase inhibitor peptide 1 and 2) were rationally designed. The primary structures of dBLIP-1 and -2 were KKGEE and KQGQE, respectively. The relations between peptide and enzyme were highly coordinated and in silico guided via the side chains of amino acid residues.
In order to set out the peptide-enzyme interactions more clearly, theoretical models of dBLIP-1 and -2 were constructed. Procheck summary of dBLIP-1 and -2 showed that, for both peptides, 100% of amino acid residues are located in the most favorable regions in the Ramachandran plot. In addition, the general qualities for the models were reliable in accordance with values -0.04 and 0.36 for the g-factor, respectively. The RMSD values and variability observed among the experimental structure templates and the modeled structure demonstrated a fold modification due to the post-modification carried out in the structure of dBLIP-1 and dBLIP-2. Moreover, neither peptide demonstrated any secondary structures. These data were confirmed by circular dichroism (data not shown). This fact was expected due to the short sequences, with only five amino acid residues in length.
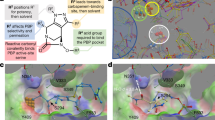
Docking analyses suggested that both peptides are able to attach to the two different β-lactamase enzymes at the center of the catalytic site, being stabilized by hydrogen bond net in the case of dBLIP-1 (Figs. 1A and 1E). As well as hydrogen bonds, electrostatic and hydrophobic interactions were also observed for dBLIP-2 (Figs. 1B and 1F). The inhibitor dBLIP-1 presented more interactions and was complementary in comparison with dBLIP-2 in the E. coli β-lactamase in silico analysis (Figure 1C). The interactions observed for dBLIP-1 were between the backbone oxygen atom (O) of lysines (Lys1 and Lys2) and the hydrogen (2HD2 and 1HD2) of Asn132 and Asn170 forming, respectively, two hydrogen bonds with distance of 2.65 and 2.5 Å. Gly3 (N and O atom) participated in two hydrogen bond interactions between Ser70 (HG) and Ala237 (HN), with distances of 2.6 and 3.1 Å, respectively. The last hydrogen bond observed was between Glu4 (OE1) and Tyr105 (HH), with distances of 3.0 Å. In contrast, the inhibitor dBLIP-2 was less interactive, presenting a hydrogen bond between the hydrogen of N atom Lys1 and oxygen atoms of Ser235 (OG) and Ala237 (O) with distance of 3.5 and 3.0 Å, respectively (Fig. 1d). Nevertheless, fewer interactions were observed in both peptides in docking analyses performed with E. coli β-lactamase (Fig. 1g). Additionally, the inhibitor dBLIP-2 showed low structural complementarity when compared to dBLIP-1 in two enzymes tested. Lysine also participates in a hydrophobic interaction, where the carbon side chain interacts with the aromatic ring of Tyr105. Another interaction observed was between the oxygen atom (OE1) of Glu5 and the nitrogen atom (NH2) of Arg244 with a distance of 3.2 Å, forming an electrostatic interaction. dBLIP-1 presented interactions between the backbone oxygen atom (O) of lysines Lys1 and the nitrogen atom (ND2) of Asn132, forming a hydrogen bond with distance of 3.45 Å. The nitrogen atom of Lys1 also participated in hydrogen bond interaction between the oxygen atom of Ser70 (OG), with a distances of 3.47 Å. The last interaction observed was between the oxygen atom of Glu5 (O) and nitrogen of the amine group of Arg244 (NH2), forming an electrostatic interaction with distances of 3.43Å. Two hydrogen bonds were observed: one between the oxygen atom (O) Lys1 and hydrogen of nitrogen atom of Asn170 (ND2), with a distance of 3.42 and the other between hydrogen of nitrogen atom of Gly3 (N) and oxygen atom (OG2) of Ser70 with a distance of 3.6 Å (Fig. 1h). Similar data were observed for other β-lactamase inhibitors10,11 in which the presence of cationic and hydrophilic residues seems to be essential for the inhibition process.

Docking studies of dBLIP-1 and dBLIP-2 and β-lactamases from two different bacterial sources.
Structural complementarity between dBLIP-1 (a) and dBLIP-2 (b) toward β-lactamase from Escherichia coli. In detail, the non-covalent interactions (dotted lines) of dBLIP-1 (c) and dBLIP-2 (D) and E. coli β-lactamase catalytic site. Structural complementarity between dBLIP-1 (a) and dBLIP-2 (b) with β-lactamase from Staphylococcus aureus. In detail, the non-covalent interactions (dotted lines) of dBLIP-1 (c) and dBLIP-2 (d) and S. aureus β-lactamase catalytic site.
Furthermore, a correlation between in silico and in vitro analyses was performed in order to better understand the mechanism of action of both peptides. In order to evaluate the in vitro results of dBLIP-1 and -2, both were synthesized by solid phase method and the identity with MALDI ToF MS analysis was checked after HPLC separation (Fig. 2a and 2b). Peptides were also synthesized for authentication and we checked that the obtained results were the same for the whole batch. dBLIP-1 was assayed against bacterial β-lactamase from Bacillus cereus 569 (Calbiochem, UK). Both peptides showed β -lactamase inhibition activity with a higher activity at 128 µg.mL−1 concentration (Fig. 2c). dBLIP-1 demonstrated greater affinity for the β-lactamase enzyme by calorimetry assays when compared to dBLIP-2. The binding affinity (K) and binding stoichiometry of both dBLIP-1 and -2 towards β-lactamase was done with isothermal titration calorimetry (ITC). The ITC results for dBLIP-1 and -2 clearly show their affinity to β-lactamase. The titration curves for the two peptides and their stoichiometry at the endpoint are shown (Fig. 2d–2f). In both cases, complexation took place, as indicated by their binding isotherm. The line through the simulated data set corresponds to the theoretical heat produced by their complex formation between β-lactamase and dBLIP-1 and -2 and the best-fit values for the parameters K, ΔH and ΔS are listed in Supplementary Table 1. The interaction processes are enthalpy-driven and based upon van der Waals interactions, along with H-bonding, because the values of ΔH and ΔS were negative12. This was also corroborated by in silico studies comparing the output energy encountered in the well-defined cluster generated after data mining for the enzyme. In addition, to improve information about interactions between dBLIPs and enzyme, the free energy values were evaluated through docking studies. It was observed that dBLIP-1 toward the β-lactamase from E. coli interactions presented -5.8 Kcal.mol−1, while the energy observed for dBLIP-2 toward the same β-lactamase was -5.4 Kcal.mol−1, demonstrating a slightly lower interaction affinity. In contrast, the energy value observed for dBLIP-1 toward the β-lactamase from S. aureus was of -4.8 Kcal.mol−1, while the energy observed for dBLIP-2 toward the same β-lactamase was -4.8 Kcal.mol−1, demonstrating an identical affinity.

Binding and inhibitory activity of Bacillus cereus 569 β-lactamase (Calbiochem, UK) by using dBLIP-1 and -2.
MALDI-ToF Mass spectra of purified dBLIP-1 (a) and dBLIP-2 (b). In vitro study for inhibition (IC50) of β-lactamase activity by dBLIP-1 and dBLIP-2, (c). ITC bonds measurement between dBLIP-1 and 2. Figures show the raw data obtained from ITC binding experiment between β-lactamase with dBLIP-1 (d) and dBLIP-2 (f). Figure representing the integration of raw heat associated data for dBLIP-1 (e) and dBLIP-2 (g). These data have been corrected by subtraction of appropriate blank experiments and then fitted with nonlinear regression; data derived after analysis have been listed in Supplementary Table 1. The binding thermodynamics for these experiments are listed in the methods section.
Since in vitro and in silico results showed that dBLIP-1 and -2 were able to interact and inhibit β-lactamase activities, the next step was obviously to challenge resistant bacteria with both peptides and see if they were able to improve the activity of β-lactam antibiotics. MIC values of amoxicillin, ampicillin and cefotaxime against β-lactamase positive clinical isolates E. coli, P. aeruginosa, S. aureus and B. cereus were varied from 128–512 µg.mL−1, showing that the isolates are clearly resistant to β-lactam antibiotics (Table 1). Moreover, when antibiotics were administered in combination with dBLIP-1 and -2, the MIC values were reduced 8–16 fold (Table 2). It is also important to note that dBLIP-1 and -2 are most active in combination with cefotaxime in comparison to other antibiotics here evaluated. Interestingly, considerable decreases in MIC values were observed for strains that are able to produce the plasmid-mediated class A β-lactamase CTX-M-14, offering initial evidence that dBLIPs may inhibit both class C and class A β-lactamases10,13.
Since a considerable reversal of bacterial resistance to β-lactams was observed here for a broad spectrum of clinical isolates and no cytotoxic effect was observed against erythrocytes and macrophages (Table 3), the efficacy of such compounds was investigated in mouse models of bacteremia and sepsis caused by Escherichia coli and also Staphylococcus aureus clinical isolates with high resistance to different antibiotics. In order to determine the in vivo efficacy of dBLIPs in a non-lethal infection by Gram-negative and -positive resistance models (Fig. 3), the CFU in the peritoneal cavity was determined.

dBLIP-1 and dBLIP-2 effects on mice in vivo model against infection of Staphylococcus aureus (top figure) and Escherichia coli (bottom figure).
Determination of CFU in S. aureus groups treated with (a) dBLIP 1 and 2, (b) penicillin (PG), (c) ampicillin (AMP) and (d) cefotaxime (CTX). Determination of CFU in E. coli groups treated with (e) dBLIP 1 and 2, (f) penicillin (PG), (g) ampicillin (AMP), (h) and gentamicin (GEM). Bars represent means and SDs from three to six independent experiments. Results are shown as mean ± SD from triplicate measurements. *p, 0.05; **p, 0.01; ***p, 0.001; comparison by ANOVA with Tukey's post hoc test.
For S. aureus, the mice were divided into groups that received different treatments in combination with commercial antibiotics: penicillin, ampicillin and cefotaxime. Comparing the negative control (which did not receive any antibiotic treatment) with the group that received dBLIP-1 and dBLIP-2, it was observed that dBLIPs alone were unable to control any bacterial infection (Fig. 3a). In the case of S. aureus infection, penicillin and ampicillin aloneat a standard dose of 100 mg.kg−1 led to a similar CFU to the untreated control. When dose was increased to 200 mg.kg−1 CFUs were slightly reduced. Furthermore, treatment with commercial antibiotics associated with dBLIP-1 and -2 led to a clear decrease in CFU count (Fig. 3b and 3c). Moreover, cefotaxime treatment led to an efficient reduction in CFU (Fig. 3d). However, when cefatoxime was associated with dBLIPs this effect clearly improved when compared to the untreated control, as previously observed in in vitro studies. For E. coli, the mice were divided into groups that received different treatments in combination with commercial antibiotics penicillin, ampicillin and gentamicin. Comparing the negative control with the group that received dBLIP-1 and –2, once more no direct bactericidal effect was observed for E. coli (Fig. 3e). Penicillin and ampicillin alone (Figs. 3f and 3g) at a standard dose of 100 mg.kg−1 did not cause any bactericidal effect. When the dose was increased to 200 mg.kg−1 a lower reduction in CFU was obtained. Treatment with commercial antibiotics associated with dBLIP-1 and -2 led to a CFU count reduction when animals were treated with ampicillin and penicillin in combination with dBLIP-1 and -2, in accordance with data obtained for Gram-positive bacteria. Gentamicine treatment, nonetheless, led to an efficient CFU decline (Fig. 3g) and both peptides only slightly improved the decrease. Similar data were obtained with other compounds and derivative fragments10,13, but dBLIPs seem to be the shortest peptides that were able to inhibit S. aureus lactamases when compared to longer proteins14. Based on these data, another question was proposed about the mechanism of action of dBLIPs. Since multifunctionality and promiscuity of short peptides have been commonly observed15, due to their ability to show different functions and to bind to different targets, are those peptides able to show a different function? Initially, dBLIPs were unable to kill bacteria by themselves, but are those peptides able to modify the mammalian immune response as host-defence peptides improving the resistance against bacteria? In order to evaluate this function, different cytokines were evaluated in mouse blood (Supplementary Fig. 1 and 2). The groups that showed a CFU increase also showed a related increase in IL-10 (Supplementary Fig. 1a and 2a), MCP-1 (Supplementary Fig. 1b and 2b) and IFN-γ (Supplementary Fig. 1c and 2c). Moreover, the values of IL-12 were different from other cytokines. For this one, the difference was that for penicillin 200 mg.kg−1 and ampicillin 100 mg.kg−1 values increased when combined with dBLIP-1. Additionally TNF-α, IL-12 and IL-6 did not show a significant difference compared to the untreated control (Supplementary Fig. 1d,e,f and 2d,e,f).
It is noteworthy that neither dBLIP showed any clear immunological response, which is extremely desirable in most designed drugs. These data suggest that dBLIPs probably do not show immune-response side effects and that deleterious activity is only related to lactamase inhibition16. In summary, dBLIP inhibitors show promise as tools to overcome resistant bacterial infections, a pervasive and growing threat to public health in several countries. They may help, as suggested in a review by Drawz et al.17, in resurrecting β-lactamase inhibitors in a world plagued by multidrug-resistant bacteria. These peptides could be useful as additives to commercial antibiotics, leading to a reduction in resistance and opening the market for novel products.
Methods
All methods described here were carried out in accordance with the approved guidelines.
Peptide designing and molecular modelling
Rational design for synthetic peptide construction was mainly based on pocket volume, which included distances between the amino acid residues that compose the β-lactamases conserved motif (KSG), Ser130 (adjacent nucleophilic amino acid) and several adjacent amino acid residues around the catalytic triad (Lys73, Ser130 and Glu166). Firstly, an isosceles trapezoid was designed inside the catalytic enzyme pocket. The distances encountered inside this geometric form showed dimensions of approximately 12 (major base) × 9 (minor base) × 13 (sides) Å in a total of 13 Å2 (Supplementary Fig. 3). Peptides were designed with ideal lengths to fit and interact with the catalytic triad and neighbouring amino acid residues localized around the catalytic site inside the trapezoid. The peptides were designed with less than five amino acid residues, flexible (presence of glycines) and soluble (presence of hydrophilic amino acid residues such as lysine and glutamic acid) (Supplementary Table 3). Moreover, the output energy that could be correlated to affinity was also detected for each in silico docking simulation with β-lactamases from Gram-negative and –positive bacteria. The parameters used to rank the best peptides to be synthesized were the energy results, with values below -4.0 Kcal.mol−1 being discarded. Five of seven constructed and analysed peptides were discarded, with four of them presenting moderate or no activity toward β-lactamases (data not shown). dBLIP-1 and -2 presented values of -5.8 and -5.4 Kcal.mol−1 toward β-lactamase from E. coli, respectively and values of -4.8 and -4.8 Kcal.mol−1 toward β-lactamase from S. aureus, respectively. The three-dimensional models for dBLIP-1 and dBLIP-2 were constructed based on the structures of 2je8 and 3pxi (pdb code), which presented 100% of identity for both peptides18,19. Fifty theoretical tridimensional peptide structures were constructed by using Modeller v.9.820,21 for each peptide. Final models were evaluated using PROCHECK for analysis of stereochemical quality. The peptide structures were visualized and investigated on Delano Scientific's PyMOL http://pymol.sourceforge.net/22. To calculate the grand average of hydropathicity, named GRAVY, ProtParam23 was used to evaluate physical-chemical parameters for a given amino acid sequence.
In silico lactamase interactions
All docking calculations were performed using the AUTODOCK 4.2 program. Docking simulations of both peptides (dBLIP-1 and dBLIP-2) were performed for two β-lactamases (EC 3.5.2.6), pdb code 1zg4 from Escherichia coli and pdb code 3blm from Staphylococcus aureus24,25. All hydrogen atoms were added by using the AutoDockTool26. Grid maps were calculated with 20 × 15 × 15 for both dBLIP-1 and -2 tested against E. coli enzyme; for the enzyme from S. aureus the grid was calculated with 35 × 35 × 15 for both dBLIP-1 and -2 and in all tests the spacing centre was 1.0 Å on the catalytic pocket of both enzymes. In order to understand the competitive in vitro inhibition observed, the simulation was optimized in a reduced region around the catalytic pocket. A Lamarckian genetic algorithm was used as the search method to find the best peptide–protein complex. Fifty docking runs were performed for each peptide, where the maximum freedom to side chains was unlocked due to length of the peptides. The generated structures were ranked in two steps: firstly a cluster with the best models with lowest free energy (below -4.0 kcal.mol−1) and secondly with a root-mean-square deviation (RMSD), for all atoms docked in the serine proteinase catalytic pocket, showing tolerance of 4Å, as recommended for blind docking. The program PyMOL http://pymol.sourceforge.net/22 was used to characterize peptide-protein interactions.
Solid phase synthesis of peptides
dBLIP-1 and dBLIP-2 were prepared by solid-phase peptide synthesis using HBTU activation procedure for Boc chemistry. Both dBLIP-1 and dBLIP-2 were synthesized from the UTMB peptide synthesis core facility (Galveston, USA) and Peptide 2.0 (USA) to authenticate the activity.
Isothermal Titration Calorimetry
To measure the binding affinity of β-lactamase with inhibitor peptides (dBLIP-1 and dBLIP-2), isothermal titration calorimetry (ITC) was performed using iTC200 Systems (GE Healthcare, USA) coupled with non-reactive Hastelloy® cells for chemical resistance. All pure samples were dissolved against phosphate buffer (pH 7.5). The same buffer degassed prior to titration and experiments were performed at 25°C. Isothermal interactions between proteins and peptides were measured by titrating over 20 injections using 40 µL peptide solution (1 mM) and proteins in sample cell with a concentration of 5 µM of 200 µL. Experiments were repeated three times.
Identification of different β-lactamase classes among clinical isolates
The identification of different β-lactamase classes present in clinical isolates was carried out following the method described by Upadhyay and co-workers (2010) as observed in Supplementary Table 2. Screening for AmpC β-lactamase production was performed by cefoxitin disk test. Isolates that yielded a zone diameter less than 18 mm (screen positive) were further subjected to confirmatory testing. The disk antagonism test was used for detection of inducible AmpC β-lactamase; cefotaxime (30 μg) and cefoxitin (30 μg) disks were placed 20 mm apart from centre to centre. Isolates showing blunting of the cefotaxime zone of inhibition adjacent to the cefoxitin disk were screened as positive for AmpC β-lactamase. The extended spectrum β-lactamase (ESBL) status of these strains was established by combined disk diffusion method per CLSI recommendations using cefotaxime (30 μg) and ceftazidime (30 μg) disks alone and in combination with clavulanic acid. Metallo β-lactamase production was detected by imipenem-EDTA disk test. Two 10 μg imipenem disks were placed on the plate and appropriate amounts of 10 μL of 0.5 M EDTA solution were added to one of them to obtain the described concentration (750 μg). The positive strain was determined if the increase in inhibition zone with imipenem and EDTA disk was ≥7 mm and then the imipenem disk alone was considered as the MBL producer.
β-Lactamase in vitro assays
The inhibition of β-lactamase activity degree by dBLIP-1 and dBLIP-2 was determined spectrophotometrically by the hydrolysis of nitrocefin as substrate. Assay mixture contained 83 mg of nitrocefin, 167 mg of BSA, 10% glycerol and 0.30 mL (0.5 μg.mL−1 of β-lactamase, obtained from Bacillus cereus 569) in a final volume of 1.5 mL of 50 mM phosphate buffer. β-lactamase activity was checked by measuring the absorbance reduction at 340 nm. Inhibitors, dBLIP-1 and dBLIP-2 at various concentrations (10 to 500 μM) were pre-incubated with the enzyme for 10 min at 30°C before addition of the substrate. Percent inhibition was calculated as 100 × [(c-r)/c], where c is the activity in control samples incubated without inhibitor and r is the remaining activity in samples incubated with inhibitor. IC50 values are calculated to inhibit 50% of enzyme activity from the plot of percent inhibition versus the logarithm value of inhibitor concentration. Kinetic parameters were derived from the initial velocity using SIGMAPLOT version 10.027.
Microorganisms
The β-lactamase positive clinical bacterial isolates used in this study (Table 2) were obtained from Priyamvada Birla Aravind Eye Hospital in Kolkata, India. The phenotypic characteristics and antibiotic sensitivity profiling of clinically isolated bacterial strains were determined by using Kirby-Bauer disk diffusion method in accordance with CLSI guidelines28. All the strains were isolated from patients associated with keratitis and species identifications were confirmed by Vitek 2 system (bioMérieux, France). Bacteria were cultured in Mueller Hinton Broth (Himedia, India) at 37°C.
Chromogenic nitrocefin β-lactamase assays in clinical isolates
Detection of β-lactamase in the clinical isolates was determined by using nitrocefin assays29 at different time intervals (1, 10 and 30 min).
Determination of minimum inhibitory concentration (MIC)
The minimum inhibitory concentrations of selected antibiotics (amoxicillin, ampicillin, cefotaxime and gentamicin) and in combination with 128 µg.mL−1 of dBLIPs against four clinical isolates were determined in Mueller Hinton Broth (MHB). Strains were characterized earlier and confirmed with 16S rDNA sequencing. Strains were pre incubated in Tryptic Soy Broth (TSB) at 30°C to achieve an optical density equivalent to 0.5 McFarland standards and used as inoculum. The MICs were determined according to CLSI guidelines28. The concentrations of each antibiotic used a range from 0.5 to 1024 µg.mL−1. All sets of experiments were repeated three times.
Cell Culture
RAW 264.7 murine macrophage-like cells were purchased from the Rio de Janeiro Cell Bank and were maintained in supplemented Dulbecco's modified eagle medium (DMEM) (Invitrogen) (4 mM L-glutamine, 10% FCS, 2 mM nonessential amino acids, 50 mg.mL−1, gentamicin and 100 units.mL−1, penicillin/streptomycin) in the presence of 5% CO2 at 37°C29.
Cell Cytotoxicity Assays
MTT assay
In order to determine the maximum non-toxic concentrations of dBLIPs and antibiotics free and in combination, cell viability was further evaluated by 3-(4,5-dimethylthiazolyl)-2,5-diphenyl-tetrazoliumbromide (MTT; Sigma) (5 mg.mL−1 in phosphate buffered saline (PBS)) in three replicates by using RAW 264.7 cells. The results were expressed as the percentage of each sample compared to the negative control (PBS buffer, pH 7.4) and positive control [mastoparan-L (1–100 μg.mL−1)]29,30,31,32.
Hemolytic assay
The hemolytic activity of free dBLIPs and combined with antibiotics was determined by using fresh mouse red blood cells (mRBCs). Control samples included adult rat serum, heat-treated (56°C, 30 min to inactivate the complement system) serum from adult rats and from neonatal rats and buffer without erythrocytes. The A415 resulting from 100% lysis was determined by analysing the supernatant of erythrocytes that had been incubated with mastoparan-L (1–100 μg.mL−1)33.
Animals
C57BL/6 mice weighing 18 to 22 g were used in this study. Animals were provided by the Central Biotery of the USP Campus in Ribeirão Preto. Animals were housed in separate cages at a constant temperature (22°C) and humidity with a 12 h light/dark cycle with ad libitum food and water. The mice were euthanized by ether inhalation or CO2 at the end of the experiments. All experiments, care and handling of animals were approved by the Ethics Committee of the Catholic University of Brasilia.
Murine non-lethal Staphylococcus aureus infection model
Mice received an intraperitoneal injection of 100 µL S. aureus (ATCC33591) 2 × 105 CFU.mL−1, previously cultured as described by Steinstraesser and co-workers34. One hour after the S. aureus injection, mice (n = 6/group) were intraperitoneally treated with dBLIP-1 and dBLIP-2 (1 mg.kg−1) in combination with penicillin (100 and 200 mg.kg−1), ampicillin (100 and 200 mg.kg−1), or cefotaxime (35 mg.kg−1). Treatments were performed every 12, 24, 48 and 72 h, for 7 days. Mice were euthanized at 7 days post infection and blood and peritoneal fluid were collected. Serial dilutions of the samples were plated in triplicate on Mannitol salt agar (Himedia, India) and the results were expressed as CFU.mL−134,35.
Murine non-lethal Escherichia coli infection model
Non-lethal bacteremia was produced in C57BL/6 mice with an i.p. inoculation of 200 μl of bacterial suspension containing 2.5 × 108 CFU of E. coli 1812446 clinical strain. One hour after the E. coli injection, mice (n = 6/group) were intraperitoneally treated with dBLIP-1 and dBLIP-2 (1 mg.kg−1) in combination with penicillin (100 and 200 mg.kg−1), ampicillin (100 and 200 mg.kg−1), or gentamicin (10 mg.kg−1). Treatments were performed every 12, 24, 48 and 72 h, for 7 days. Mice were euthanized at 7 days post infection and blood and peritoneal fluid were collected. Serial dilutions of the samples were plated in triplicate on Mueller Hinton Agar (Himedia, India) plates. The plates were incubated overnight at 37°C and the bacterial colonies were counted on the following day. The bacterial limit count detected by this method was 100 CFU.ml−136.
Cytokine assay
Cytokines IL-6, IL-10, IL-12p70, MCP-1, IFN-γ and TNF-α were measured in plasma of mice subjected to the non-lethal S. aureus and E. coli infection model, using an ELISA kit (Peprotech, USA) according to the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± SD of all samples. Statistical significance of fatality rates between different groups was performed by Kaplan–Meier test. The other data were submitted to one-way analysis of variance (ANOVA) followed by Bonferroni test. Values of p < 0.05 were considered statistically significant. GraphPad Prism software v5.0 (GraphPad Software, USA) was used for all statistical analyses.
References
Levy, S. B. & Marshall, B. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 10, 122–129 (2004).
Sandora, T. J. & Goldmann, D. A. Preventing lethal hospital outbreaks of antibiotic-resistant bacteria. N Engl. J. Med. 367, 2168–70 (2012).
Turner, C. E. et al. Molecular analysis of an outbreak of lethal postpartum sepsis caused by Streptococcus pyogenes. J Clin Microbiol. 51, 2089–95 (2013).
Tse, H. et al. Molecular characterization of the 2011 Hong Kong scarlet fever outbreak. J. Infect Dis. 206, 341–51 (2012).
Walker, M. J. & Beatson, S. A. Epidemiology. Outsmarting outbreaks. Science 338, 1161–1162 (2012).
Sarraf-Yazdi, S. et al. A 9-Year retrospective review of antibiotic cycling in a surgical intensive care unit. J. Surg. Res. 176, e73–8 (2012).
Tenover, F. C. & Hughes, J. M. The challenges of emerging infectious diseases. Development and spread of multiply-resistant bacterial pathogens. JAMA 275, 300–304 (1996).
Jahnsen, R. D. et al. Antimicrobial activity of peptidomimetics against multidrug-resistant Escherichia coli: a comparative study of different backbones. J. Med. Chem. 55, 7253–61 (2012).
Gary, W. et al. Binding properties of a peptide derived from lactamase inhibitory protein. Ant. Agents Chem. 45, 3279–3286 (2001).
Eidam, O. et al. Fragment-guided design of subnanomolar β-lactamase inhibitors active in vivo. Proc Natl Acad Sci U S A. 109, 17448–53 (2012).
Rodkey, E. A. et al. Crystal structure of a preacylation complex of the β-lactamase inhibitor sulbactam bound to a sulfenamide bond-containing thiol-β-lactamase. J Am Chem Soc. 134, 16798–804 (2012).
Wang, S. H. et al. Thermodynamic analysis of the molecular interactions between amyloid beta-peptide 42 and (-)-epigallocatechin-3-gallate. J Phys Chem B. 114, 11576–11583 (2010).
Eidam, O. et al. Design, synthesis, crystal structures and antimicrobial activity of sulfonamide boronic acids as beta-lactamase inhibitors. J Med Chem 53, 7852–7863 (2010).
Yuan, J. et al. Identification of a β-lactamase inhibitory protein variant that is a potent inhibitor of Staphylococcus PC1 β-lactamase. J Mol Biol. 406, 730–44 (2011).
Franco, O. L. Peptide promiscuity: an evolutionary concept for plant defense. FEBS Lett. 585, 995–1000 (2011).
Ripoll, A. In vitro selection of variants resistant to beta-lactams plus beta-lactamase inhibitors in CTX-M beta-lactamases: predicting the in vivo scenario? Antimicrob Agents Chemother. 55, 4530–4536 (2011).
Drawz, S. M. New β-lactamase inhibitors: a therapeutic renaissance in an "MDR world. Antimicrob Agents Chemother 58, 1835–46 (2014).
Tailford, L. E. et al. Mannose foraging by Bacteroides thetaiotaomicron: structure and specificity of the beta-mannosidase, BtMan2A. J Biol Chem 282, 11291–11299 (2007).
Wang, F. et al. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 471, 331–335 (2011).
Eswar, N. et al. [Comparative protein structure modeling using Modeller, Chapter 5]. [Curr Protoc Bioinformatics] [5-6]. (John Wiley and Sons, London, 2006).
Sumathi, K. et al. 3dSS: 3D structural superposition. Nucleic Acids Res 34, W128–132 (2006).
Lano, D. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA, USA 1 (2002).
Wilkins, M. R. et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112, 531–552 (1999).
Stec, B. et al. Structure of the wild-type TEM-1 beta-lactamase at 1.55 A and the mutant enzyme Ser70Ala at 2.1 A suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Crystallogr D Biol Crystallogr 61, 1072–1079 (2005).
Herzberg, O. Refined crystal structure of beta-lactamase from Staphylococcus aureus PC1 at 2.0 A resolution. J Mol Biol 217, 701–719 (1991).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31, 455–461 (2010).
Perez-Llarena, F. et al. The bla gene of the cephamycin cluster of Streptomyces clavuligerus encodes a class A beta-lactamase of low enzymatic activity. J Bacteriol 179, 6035–6040 (1997).
Institute, C. a. L. S. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard-eighth edition. CLSI document M11-A7. (2007).
O'Callaghan, C. H. et al. Novel method for detection of beta-lactamases by using a chromogenic cephalosporin substrate. Antimicrob Agents Chemother 1, 283–288 (1972).
Lopez-Abarrategui, C. et al. Functional characterization of a synthetic hydrophilic antifungal peptide derived from the marine snail Cenchritis muricatus. Biochimie 94, 968–974 (2012).
Pasupuleti, M. et al. End-tagging of ultra-short antimicrobial peptides by W/F stretches to facilitate bacterial killing. PLoS One 4, e5285 (2009).
Laakkonen, P. et al. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad Sci U S A 101, 9381–9386 (2004).
Jasinski, M. et al. A novel mechanism of complement-independent clearance of red cells deficient in glycosyl phosphatidylinositol-linked proteins. Blood 103, 2827–2834 (2004).
Steinstraesser, L. et al. Innate defense regulator peptide 1018 in wound healing and wound infection. PloS One 7, e39373 (2012).
Jacobsen, F. et al. Activity of histone H1.2 in infected burn wounds. J Antimic Chem 55, 735–741 (2005).
Li, S. A., Lee, W. H. & Zhang, Y. Efficacy of OH-CATH30 and its analogs against drug-resistant bacteria in vitro and in mouse models. Antimicrob Agents Chemother 56, 3309–3317 (2012).
Acknowledgements
The authors would like to thank technician Tania Paula Garcez de Lucena Santana for bioassay facilities for animal care. The authors also thank Dr. Jayangshu Sengupta and Dr. Suman Saha, Priyamvada Birla Aravind Eye Hospital, 10, Loudon Street, Kolkata-700 017, W B, India, for providing the necessary help to study clinical isolates. This work was supported by CNPq, CAPES, FAPDF and UCB.
Author information
Authors and Affiliations
Contributions
S.M.M., L.M., O.N.S. and O.L.F. designed the experiments. L.M. performed in silico analyses. O.N.S., S.C.D. and C.F.Jr. performed anti-bacterial and immunological analyses. O.N.S. and I.C.M.F. performed in vivo analyses. S.M.M., A.B. and T.K.H. performed calorimetry and mass spectrometry analyses. S.M.M., L.M., O.N.S. and O.L.F. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Mandal, S., Migliolo, L., Silva, O. et al. Controlling resistant bacteria with a novel class of β-lactamase inhibitor peptides: from rational design to in vivo analyses. Sci Rep 4, 6015 (2014). https://doi.org/10.1038/srep06015
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06015
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
