
Abstract
Glaucoma is a chronic disease that causes structural and functional damage to retinal ganglion cells (RGC). The currently employed therapeutic options are not sufficient to prevent vision loss in patients with glaucoma; therefore, there is a need to develop novel therapies, which requires the creation of functional, repeatable and easy-to-utilize animal models for use in pre-clinical studies. The currently available models ensure only low to moderate damage in optic nerves, with high variation in the outcomes and poor repeatability. We have developed an effective and reproducible rat glaucoma model based on a previous idea for a “Bead Model” in mice, which could be useful in future glaucoma research. Additionally, in an attempt to achieve rapid elevation of Intraocular Pressure (IOP), we included an initial “high-pressure injury” as part of this method, which serves as the equivalent of a severe glaucoma attack. These modifications made it possible to achieve longer lasting IOP elevation with chronic damage of retinal ganglion cells.
Similar content being viewed by others

Introduction
Glaucoma is a chronic disease that causes both structural and functional damage to retinal ganglion cells (RGC) and their axons, leading to irreversible blindness. It has been estimated that, in global terms, glaucoma is second most common cause of blindness; currently affecting more than 60 million people1. The exact mechanism of axonal damage is still unknown. Previously, it was thought that glaucoma was caused only by increased intraocular pressure (IOP); however, it is now known that although IOP is important, it is not the only pathological factor2,3. There are other factors believed to be involved in the development of glaucomatous neuropathy, such as inflammatory processes, oxidative stress, metabolic abnormalities and blood flow disturbances4,5,6. The currently employed therapeutic options are not sufficient to prevent vision loss in glaucoma patients; therefore, there is a need to develop novel therapies that can protect retinal ganglion cells from degeneration7,8,9.
New therapeutic strategies for glaucoma will require the development of functional, repeatable, easy-to-utilize, low-cost animal models for use in pre-clinical studies. Additionally, these models should mimic conditions that appear during the course of human glaucoma. The models should also shed light on the mechanisms underlying the death of RGCs, not simply on the IOP lowering effect of commercially available drugs, as is the case for the majority animal models of glaucoma that are currently used.
Artificially induced elevated IOP in animals is a widely used experimental approach to mimic human ocular hypertension and it has been shown to lead to significant RGC loss and optic nerve damage10. Rodents are the most popular animals in glaucoma models, with mice and rats being the most commonly used11. The predominance of rats is based on their similarities with humans with respect to the anatomical and developmental features of the ocular anterior segment, aqueous humor circulation and optic nerve changes caused by increased IOP12. There are different IOP-based animal models of ocular hypertension; however, they evoke different mechanisms of RGC loss and axonal damage13. To be classified as glaucomatous, the damage induced in the retina and optic nerve should be sufficiently distinct to allow an estimation of the putative effectiveness of the applied treatment strategies but slow enough to resemble the chronic character of true glaucoma.
It is clinically known that patients with a history of acute glaucoma attacks and highly increased IOP tend to develop glaucomatous neuropathy more rapidly than patients with chronic, intermediate IOP elevation. It is the abrupt increase in IOP that has been recognized as a critical stress factor for RGCs and their axons2,3,14,15,16.
We have improved the previously described mouse “Bead Model” originally devised by Cone et al. and adapted it for rats10. Additionally, in an attempt to achieve an abrupt elevation of IOP, we added an initial “high-pressure injury” to this method; this injury is the equivalent of a severe glaucoma attack. These modifications made it possible to achieve longer lasting IOP elevation with chronic damage, as reflected in the loss of retinal ganglion cell bodies and optic nerve axons. The model was evaluated with regular IOP measurements and fundus photographs as well as with different post mortem tissue analyses.
Results
Our glaucoma model is based on an intracameral injection of polystyrene microbeads in a viscoelastic suspension (5 μl of viscoelastic solution, 5 μl of beads with a particle diameter of 6.0 μm and 5 μl of beads with a particle diameter of 10.0 μm). The suspension of beads was injected rapidly through a glass needle, which generated an initial high pressure in the anterior chamber that was responsible for visible corneal edema, proper bead distribution and increased IOP levels as well as RGC loss. In contrast to described method, slow injection of the corresponding bead suspension as well as injection of beads with different diameters (1.0 and 6.0 μm) were performed during model development. Slow injection due to poor needle emptying resulted in a lower initial IOP (with no corneal edema); similarly, smaller bead sizes resulted in lower, unstable IOPs and a low rate of RGC loss.
Mean IOP
The mean 6-week IOP in eyes with induced ocular hypertension (after bead injection with initial high-pressure injury) was 30.9 ± 3.2 mmHg, whereas this value was 11.4 ± 0.8 mmHg in healthy control eyes. At each measurement time point (days 1, 7, 14, 21, 28, 35 and 42) as well as the mean 6-week IOP in bead-injected eyes was significantly higher compared with that in healthy eyes (p < 0.05, Wilcoxon paired test) (Figure 1).

Mean IOP values in eyes after bead injection and in healthy control eyes.
Immediately after injection of beads, the IOP increases rapidly and corneal edema appears when the IOP exceeds the level of swelling pressure (≈55 mmHg). The IOP reaches a maximum immediately prior to removal of the glass needle from the anterior chamber (approximately 3 minutes after the start of the injection). After the needle has been removed, IOP rapidly declines due to fluid outflow from the anterior chamber. When IOP decreases below the level of swelling pressure, it creates the conditions needed for restoration of corneal transparency (this takes approximately 6 hours). During first 24 hours, when the corneal wound is healing, IOP oscillates around an intermediate level. After the corneal wound has sealed, IOP increases, due to the impaired aqueous humor trabecular outflow and it reaches a maximum level after 7 days. The dynamic rebuilding of the trabecular meshwork causes a slow decline in the IOP value, although it remains at a significantly elevated level for the entire experiment (a period of 6 weeks).
Fundus photographs
In the eye fundus, we observed features typical for glaucomatous damage, including optic nerve disc cupping (the cup/disc ratio in glaucomatous eyes had increased to approximately 0.55 ± 0.05 compared with 0.34 ± 0.025 in healthy eyes; p < 0.05, U-Mann Whitney test) and localized retinal atrophy (Figure 2 A–D).

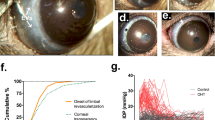
Anterior segment photographs in strong, coaxial bright light.
In healthy eyes only, the reflex from the corneal surface is visible (A). Except for the area of the corneal reflex, it is possible to observe the localization of the beads after bead injection (B). At 6 weeks after intracameral bead injection, the beads were localized in the corneal endothelium (asterisk, *), on the iris surface (cross, +) and in the iridocorneal angle (arrows), especially in the inferior area. In fundus photographs, optic nerve disc cupping is visible, with an increased c/d ratio in glaucomatous eyes (D) compared with healthy eyes (C). E-hematoxylin and eosin-stained cross-sections of the iridocorneal angle with tightly packed beads of two different sizes (the large arrow marks a 10.0-μm diameter bead and the small arrow marks a 6.0-μm diameter bead). Scale bars = 500 μm (left) and 50 μm (right).
Loss of retinal ganglion cells and optic nerve axons
The mean 6-week loss of cell bodies in the retinal ganglion cell layer was approximately 34% for β3-tubulin-positive cells, 29.2% for NeuN-positive cells and 20% for DAPI-positive cells. The numbers of cells that were positive for each marker were significantly lower in the glaucoma group than in the healthy controls (p < 0.05, Wilcoxon paired test). In healthy eyes, it was estimated that 7.9 ± 0.6, 4.8 ± 0.6 and 3.6 ± 0.4 cells per 200 μm of each retinal section length were positive for DAPI, NeuN and β3-tubulin; in glaucomatous eyes, these numbers were 6.4 ± 0.7, 3.4 ± 0.5 and 2.4 ± 0.5, respectively. The mean 6-week loss of optic nerve axons was approximately 28%. The estimated total number of axons (102413 ± 10180 in the healthy group and 73069 ± 14339 in the glaucoma group) as well as the axon density/μm2 (0.46 ± 0.06 in the healthy group and 0.35 ± 0.07 in the glaucoma group) was significantly lower in the glaucoma group (p < 0.05, Wilcoxon paired test) (Figure 3 A–C). There was a clear significant correlation observed between IOP values and RGC cell bodies as well as axon numbers (r = −0.8, p < 0.05 and r = −0.7, p < 0.05, respectively; Spearman correlation rank test).

Mean number of cells in the ganglion cell layer of the retina (A), total number of axons in the optic nerve (B) and optic nerve axon density (C) in healthy and glaucomatous eyes (after 6 weeks; rapid beads injection with initial high-pressure injury). (D) – mean number of cells in the ganglion cell layer of the healthy retina and 7 days after beads injection. Immunostaining revealed no visible difference between beads-injected and healthy retinas for β3-tubulin (E, F), however there was slightly increased number of TUNEL positive cells within ganglion and outer nuclear layer in beads-injected retinas ((G), (H), arrows). The lowest panel shows a comparison between the groups used in the optimization of the model. The combination of bead sizes as well as the use of an initial “pressure injury” resulted in higher mean IOP values (I) and in an efficient and reproducible level of retinal ganglion cell loss (J). The asterisk indicates statistical significance, ns – not significance. Scale bar = = 100 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
To exclude presence of early ischemic retinal damage, caused by beads injection and initial high-pressure injury, group of 5 animals was sacrificed one week after beads injection and eyeballs cross sections were processed for immunostainings and RGCs counting. Although, the number of β3-tubulin positive RGCs in bead-injected eyes within this group (3.5 ± 0.3 per 200 μm) was slightly lower when compared with RGCs number in control contralateral eyes (3.7 ± 0.5), this cell loss (approximately 5%) was not significant (p > 0.05, Wilcoxon paired test; Figure 3 D). Immunostaining against β3-tubulin revealed no visible difference between beads-injected and healthy retinas, however there was slightly increased number of TUNEL positive cells within ganglion and outer nuclear layer in retinas from beads-injected eyes (Figure 3 E–H).
Model development
During model optimization, we first tested two different bead sizes. First, a group of 23 male Wistar rats (8 weeks old) were injected with 15 μl of a mixture of viscoelastic solution and beads with particle diameters of 1.0 and 6.0 μm. Injection was performed without generation of an initial high-pressure injury. The IOP achieved with this combination–17.3 ± 1.7 mmHg–caused an 18.3% loss of RGC bodies during 6 weeks of observation (estimated for β3-tubulin-positive cells). Another group of 20 male Wistar rats (8 weeks old) were injected similarly with a suspension of beads (15 μl of a viscoelastic suspension containing beads with particle diameters of 6.0 and 10.0 μm), but without generating an initial high-pressure injury (slow bead injection, without corneal edema). The mean IOP obtained under these conditions was 25.3 ± 8.3 mmHg, which evoked a 22.3% loss of RGC bodies during 6 weeks (Figure 3 I, J).
In summary, the method described here (15 μl of a viscoelastic suspension containing beads with particle diameters of 6.0 and 10.0 μm, with an initial high-pressure injury), was considered to be the most effective; thus, it was repeated in eighteen 42-week-old Wistar rats. In this group, the mean IOP generated was 31.1 ± 5.4 mmHg, which led to a mean RGC loss of approximately 34.6% and a mean axon loss of 25%. These results are evidence of the good reproducibility of the method (detailed data not shown) (Figure 3 E).
Immunohistochemistry
Using hematoxylin-eosin staining, the beads could be visualized in structures of the iridocorneal angle, deeply packed between the cornea and iris; both larger (big arrow) and smaller (small arrow) bead particles were observed (Figure 2 E). In whole mounted retinas, glaucomatous damage was visible in reduced numbers of cells that were positive for β3-tubulin (RGC marker) and NeuN (neuronal nuclei marker) (Figure 4 A–D). The loss of cells in the ganglion cell layer was also visible in the retinal cross sections (Figure 4 E–H). In all types of specimens (retinas and optic nerves) the typical signs of glaucomatous neuropathic damage (i.e., activation and proliferation of glial cells) could be observed (Figure 4 I–N). The elevated IOP induced cellular apoptosis mostly in ganglion cells and slightly in the inner nuclear layer (Figure 4 O–P). In sections of optic nerves stained with 2% toluidine blue, sectorial damage with fibrosis, axonal swelling and phagocytic infiltration could be discerned (Figure 5).

Immunofluorescence staining of healthy and glaucomatous retinas.
β3-tubulin/NeuN-positive cell loss was visible in both whole mounted retinas ((A,B): 10× magnification, scale bar = 500 μm; (C,D): 20× magnification, scale bar = 50 μm) and in retinal cross-sections ((E–H), scale bar = 100 μm). In addition, thinning of retinal ganglion cell axons can be observed ((A–D), arrows). (I–N): Immunofluorescence staining of retinas and optic nerves revealed activation and proliferation of glial cells as a response to glaucomatous damage ((I,J): whole mounted retinas, scale bar = 500 μm; (K,L): retinal cross-sections, scale bar = 100 μm; (M,N): optic nerve cross-sections, scale bar = 500 μm). (O,P): TUNEL-fluorescent staining of retinal cross-sections from healthy (O) and glaucomatous (P) eyes. In healthy retinas, TUNEL-positive cells can be observed mostly within the ONL and single cells can be observed in the GCL. In glaucomatous retinas, there was a slightly increased number of TUNEL-positive cells in the ONL as well as some cells in the INL; scale bar = 100 μm. GCL, ganglion cells layer; INL, inner nuclear layer; ONL, outer nuclear layer. The graph miniatures present the number of cells in the GCL specific for DAPI, NeuN and β3-tubulin.

Healthy (A) and glaucomatous (B) optic nerve cross-sections stained with 2% toluidine blue, revealing sectorial damage. Areas of glaucomatous damage are marked with arrows (B); scale bar = 500 μm. (C,D): High magnification microscopy of healthy (C) and glaucomatous (D) optic nerves revealed multiple neuropathic features, including (D) axon swelling (cross, #), phagocyte infiltration (arrow) and fields with gliosis (asterisk, *); scale bar = 10 μm. The graphs present the number and density of optic nerve axons.
Discussion
There are many different animal models of inducible glaucoma; these can be subdivided according to the mechanism of IOP elevation into pre-trabecular, trabecular and post-trabecular12. Pre-trabecular models are represented by intracameral injections of different combinations and types of microbeads (polystyrene, magnetic), while the trabecular model undergoes laser trabeculoplasty, which is difficult to perform due to the small size of the rodent eye. Post-trabecular models involve various treatments of the episcleral veins, such as laser coagulation, cauterization or hypertonic saline injection. Post-trabecular models achieve a rapid but short-lasting elevation of IOP, although they have many disadvantages17. Laser-induced damage to the veins can also injure ciliary nerves and episcleral vein occlusion can evoke local ischemic and hypoxic changes in the corneal limbus as well as in the choroid, accompanied by a risk of secondary neovascularization. These processes may radically alter the cascade of biochemical reactions, leading to cell death18. Pre-trabecular and trabecular models achieve minimal ocular damage compared with their post-trabecular counterparts and furthermore, they display the best agreement with the actual course of glaucoma. However, all of these techniques are characterized by high variability and low repeatability. Thus, there is a clear need to improve these models in experimental glaucoma research10.
Table 1 lists the available reports that describe the use of the “Bead Model” in rodents to achieve IOP elevation. Most of these studies were conducted on mice and only low to moderate damage was achieved in optic nerves with high variations in the outcomes10,19,20,21,22. Even when the “Bead Model” was employed using a 4 + 1 protocol (4 μl of beads + 1 μl of viscoelastic solution), which has been reported as being the least variable and the most reproducible protocol, there was still extensive variation in the observed results. Moreover, the data obtained appeared to be incomplete or not sufficiently accurate, leading to difficulties in evaluation and comparison with other studies20,23,24.
When rats were used, the main problem encountered with the polystyrene “Bead Model” was the short duration of IOP elevation (up to 2 weeks). This does not cause enough damage to the cells despite the satisfactory IOP elevation22. Samsel et al. proposed a novel “Bead Model” in which magnetic particles could cause highly elevated IOP values and showed that this resulted in severe axon damage within 4 weeks. The short duration and severe damage evoked by this model can be classified as acute neuropathy and not chronic glaucomatous damage, although IOP elevation is the basic mechanism through which the nerves are injured25,26,27.
It is important to note that the moderately, chronically elevated IOP in clinical glaucoma evokes significant, irreversible damage in human eyes over a time period of years. Untreated glaucoma requires approximately 14.4, 6.5 and 2.9 years to progress from early-stage to end-stage if the IOP is in the range of 21 to 25 mmHg, 25 to 30 mmHg and more than 30 mmHg, respectively28,29,30,31,32,33. The short lifespan of the small rodents being utilized in experimental glaucoma models provides a relatively short time to follow up on pressure-induced changes. For this reason, only mild IOP elevation is achieved. The combination of mild IOP elevation together with the relatively short time frame in which experiments can be conducted means that it is not possible to inflict sufficient cellular/axonal loss to match that encountered in clinical glaucoma. To compensate for this, a primary injury is often induced, which will initiate a cascade of events leading to cell death; however, the IOP must be simultaneously elevated to properly mimic glaucoma. A similar mechanism may be functioning in human glaucoma. Indeed, the abrupt appearance of highly elevated IOP is one of the known factors that can accelerate glaucomatous neuropathy progress, as has been observed in human patients suffering from even a single attack of acute glaucoma2,14,16.
The rat “Bead Model” developed here makes it possible to generate moderate, chronic IOP elevation that lasts for at least 6 weeks (longer time periods have not been examined). The initial high-pressure injury is a crucial factor because it prolongs the duration of the stable but intense IOP elevation at a level that is sufficient to inflict retinal and optic nerve damage. Because the initial, highly elevated IOP lasts only for a short time (i.e., the first few minutes after bead injection), it does not cause severe damage signs in retinal histology. Rather, initial high-pressure injury is most likely attributable to the improved distribution of beads in the anterior chamber and initiation of the pressure-related cellular death cascades, which will subsequently lead to degeneration due to presence of chronically, moderately elevated IOP. This improved glaucoma model is useful for in vivo monitoring at many stages (initial pressure elevation-corneal edema, bead distribution, fundus photographs, IOP measurements), which is helpful in predicting the final outcome. We showed the features of glaucomatous damage using tissue histology and we were able to achieve similar results in repeat experiments. Previously, Hernandez et al reported that “layer-localized” cellular damage was not associated with the death of any specific type of cell, including neurons41. Moreover, these authors showed that ocular hypertension can lead to the degeneration of subpopulations of amacrine cells as a secondary result of RGC death and neuronal synaptic dysregulation in a rat model. Similarly, we observed a slightly increased number of TUNEL-positive apoptotic cells in the INL of glaucomatous retinas. However, the possibility that some of the positive staining might represent microglia or Müller cells cannot be excluded.
The presence of beads in the anterior chamber can be considered as one disadvantage of this model because they can impair visualization of the eye fundus; however, it is possible to select animals with restored transparency after the beads have spread deeply into the iridocorneal angle.
This modified glaucoma “Bead Model” makes it possible to achieve both long-lasting, significantly elevated IOP and chronic glaucomatous damage to the retina and optic nerve. The elevation of IOP at an early time point (e.g., via a pressure injury) increases the effectiveness of IOP elevation and the loss of cells/axons. This experimental rat glaucoma “Bead Model” with an initial high-pressure injury was found to be a rapid and reproducible method with a low level of variation that we predict will be useful in future glaucoma research.
Methods
Equipment and reagents
-
a
25 μl Hamilton syringe with iron needle
-
b
Plastic tubing that can be attached to the Hamilton needle
-
c
Microelectrode puller (P1000 Flaming/Brown, Harvard Apparatus, Canada)
-
d
Glass capillaries, 0.86 mm in diameter (World Precision Instruments, Sarasota, FL, US)
-
e
Laboratory tonometer (TonoLab, Icare, Finland)
-
f
Polystyrene microbeads with particle diameters of 10.0 μm and 6.0 μm (Polysciences, Warrington, PA, US)
-
g
Low-viscosity immersion oil (Cargille Laboratories, Cedar Grove, NJ, US)
-
h
1.4% viscoelastic solution (Microvisc Plus, Bohus Biotech, Sweden)
-
i
Topical anesthesia (Proxymetacaine Hydrochloride, 0.5%, Alcaine, Alcon, Fort Worth, TX, US) or other preparations containing proxymetacaine
-
j
General anesthesia (ketamine (VetaKetam) 100 mg/ml, VET-AGRO, Poland and xylazine (Xylapan) 20 mg/ml, Vetoquinol Biowet, Poland)
-
k
Antibiotic ointment
Animals
All animals were provided by the Animal House of the Experimental Medicine Center, Medical University of Silesia, Katowice, Poland and were treated in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the EC Directive 86/609/EEC for animal experiments using the protocols approved and monitored by the Local Committee for Animal Experiments of the Medical University of Silesia. For glaucoma induction, we used 8-week-old male Wistar rats weighing approximately 200 g. The right eye of each animal was the target of the experimental procedures; the left eye was maintained as a healthy control.
To elevate the IOP, we used an intracameral injection of a 2.5% polystyrene microbead suspension (Polysciences, Warrington, PA, US) in phosphate buffered saline (PBS) with an approximate density of 2 × 108 beads/ml. During model optimization, we compared the different suspension volumes and bead sizes used for IOP elevation and we found that the combination of 5 μl of polystyrene microspheres with a particle diameter of 10.0 μm and 5 μl of microspheres with a particle diameter of 6.0 μm was most efficient for obtaining a highly and constantly elevated IOP in rats.
Preparation of animals
On the day of glaucoma induction (n = 25 rats), IOP measurements were performed in both eyes with a laboratory rebound tonometer (TonoLab, Icare, Finland). Rebound tonometry is based on the measuring principle, in which a very light probe is used and rebounds against the corneal surface to make fleeting contact. The resilience of the rebound of this probe is processed into the IOP value. After IOP measurements, the animals received general anesthesia (intraperitoneal injection of 50 mg/kg ketamine (VetaKetam 100 mg/ml), VET-AGRO, Poland and 5 mg/kg xylazine (Xylapan 20 mg/ml), Vetoquinol Biowet, Poland) and topical anesthesia (proxymetacaine hydrochloride (Alcaine), Alcon, Fort Worth, TX, US) applied to the corneal surface [TROUBLESHOOTING 1].
Preparation of injection set
A glass microneedle with a tip diameter of approximately 50 μm was prepared using a microelectrode puller and combined with a 22-G needle connected to a 5 ml syringe using plastic tubing. The syringe contained low-viscosity microscope immersion oil (Cargille Laboratories, Cedar Grove, NJ, US), which was used to fill the tubing and glass needle. We observed that the use of low-viscosity oil improved the filling of the microneedle with beads. When the whole system had been filled with oil, the syringe with its needle was removed and the tubing was connected to the 22-G needle of a 25-μl Hamilton syringe. After the syringe was changed, the glass capillary was filled with 5 μl of 1.4% viscoelastic solution (Microvisc Plus, Bohus Biotech, Sweden). The use of a high-density viscoelastic solution (1.4% instead of 1.0%) resulted in better IOP values; this was related to better sealing of the wound after injection and the reduced outflow of the suspension injected into the anterior chamber (AC). After the viscoelastic solution, capillary was filled with 5 μl of polystyrene microspheres with a particle diameter of 10.0 μm followed by 5 μl of microspheres with a particle diameter of 6.0 μm (Figure 6 A). This arrangement made it possible to inject the smaller beads initially, followed by the larger size particles, which ensured better penetration into the iridocorneal angle and trabecular meshwork. The role of the viscoelastic solution was to minimize the dead space in the needle as well as to close the corneal wound after injection. The total volume injected was 15 μl [TROUBLESHOOTING 2, 3 and 4]. Before being drawn into the needle, the beads were washed once in absolute ethanol, centrifuged for 1 minute at 1000 rpm and washed three times in PBS.

Modified “Bead Glaucoma Model” in Wistar rats.
(A): Glass needle filled with a suspension of viscoelastic solution, 10 μm beads (blue) and 6 μm beads (yellow). (B,C): Preparation and IOP measurement. (D,E): Immobilization of the eyeball with microforceps. (F): Introducing the glass needle into the anterior chamber at an approximately 45 degree angle to the corneal surface. (G): Injection of beads, slight corneal edema visible. (H,I): Schematic differences between low- and high-speed bead injection. (H): With low-speed injection, the beads formed a clot in the center of the anterior chamber (AC), impairing the distribution into the iridocorneal angle. The final outcome is that there is no separation of the beads. (I): With rapid injection, high pressure, which is generated in the AC, pushes the beads deeply into the iridocorneal angle. The limited volume of the AC and the high pressure improve the separation of beads and their distribution, which results in higher values of IOP.
Injection procedure
The topically anesthetized lower eyeball conjunctiva was grabbed with microforceps to immobilize the eyeball. A glass microneedle was introduced into the anterior chamber near the corneal limbus at an angle of approximately 45 degrees to the corneal surface in the lower quadrant of the cornea, avoiding contact with the lens anterior capsule, iris and corneal endothelium [TROUBLESHOOTING 5 and 6]. When the needle tip had passed through the cornea, the entire volume was injected rapidly (duration of approximately 5 seconds) into the anterior chamber. We observed that rapid injection achieved much better needle emptying and better distribution of beads in the AC than slow injection (Figure 6 B–I). This fast injection resulted in higher IOP levels and more efficient loss of RGCs. As a marker of successful injection, a transient peripheral corneal edema should appear during injection of the beads as a result of the initially highly elevated IOP [TROUBLESHOOTING 7]. The edema disappeared in the first 6 hours after the procedure. To minimize the outflow of beads through the wound and also to maintain the initially highly elevated IOP level, the needle was kept in the anterior chamber for approximately 3 minutes after injection and antibiotic ointment was applied onto the corneal surface (ofloxacin, 0.3% (Floxal), Bausch & Lomb, Rochester, NY, US) [TROUBLESHOOTING 8 and 9].
Because of the small size of the rat eye and the small suspension volume used for IOP elevation, it is particularly important to deliver as many beads as possible into the anterior chamber. It is essential to carefully control each step of current protocol to achieve satisfactory IOP values.
Troubleshooting
TROUBLESHOOTING 1. Local anesthesia is used to reduce pain-related mydriasis, which improves penetration of the beads into the iridocorneal angle. Proxymetacaine also has vasoconstrictive properties, which can reduce the risk of bleeding from the conjunctiva and iris.
TROUBLESHOOTING 2. The glass needle filled with beads tended to dry out, which may block the lumen of the needle. For this reason, after filling, the needle should be wrapped with a cloth soaked with 70% ethanol if it will not be in use for longer than 5 minutes.
TROUBLESHOOTING 3. Beads that remained in the needle after injection tended to dry out and stick to the walls, which reduced the needle capacity on its next filling. Although the needle can be used multiple times, it needs to be washed between injections with 70% ethanol and subsequently rinsed with sterile water.
TROUBLESHOOTING 4. The presence of air bubbles in the immersion oil impaired needle filling due to the poor compressibility of air. Any oil containing air bubbles should always be replaced with fresh solution.
TROUBLESHOOTING 5. The small size of the pupil increases the risk of iris injury from the needle tip; however, on the other hand, it improves penetration of the beads by opening the iridocorneal angle. Pupil dilation might disturb the optimal distribution of the beads and furthermore, the injected suspension may penetrate into the posterior chamber of the eye.
TROUBLESHOOTING 6. Flat insertion of the needle into the anterior chamber (with a small angle) poses a risk of injecting beads intrastromally into the cornea; upright insertion into the anterior chamber (with large angle) increases the risk of iris and lens injury with the needle tip.
TROUBLESHOOTING 7. When the entire volume of the needle had been injected into the anterior chamber, it is advisable to release the conjunctival forceps because this minimizes pressure on the eyeball and reduces outflow of the beads.
TROUBLESHOOTING 8. Antibiotic ointment should be applied on the corneal surface after outflow of the injected fluid through the corneal wound has completely stopped. Application of the ointment too early may re-open the wound due to pressure on the cornea and this may “squeeze” the beads out of the anterior chamber.
TROUBLESHOOTING 9. There are many colors of beads available. In our experience, it is better to avoid white and red/purple beads, as they may make it difficult to discern bleeding or the presence of inflammation in the anterior chamber after injection.
In vivo methods for model evaluation
Corneal edema
Corneal transparency observation, which is based on the theory of a corneal endothelium barrier, was used as a tool to confirm the initial high IOP elevation, which determines the pressure-related injury. In healthy eyes, the corneal endothelial cell layer represents a barrier for aqueous humor inflow from the anterior chamber to the corneal stroma. Corneal edema is prevented by both the physical barrier of the endothelium and by the active mechanism of the Na+/K+/ATPase pump34. In the clinic, IOP-related corneal edema is observed as a severe glaucoma attack, which is one of the diagnostic criteria for severe glaucoma. The mechanism of IOP-related corneal edema has been widely described in animal models35,36. The corneal swelling properties are characterized by the following: corneal swelling pressure (SP), which is related to the properties of stromal glycosaminoglycans, their ability to absorb and retain a large amount of fluid, limit the pressure of fluid that can be maintained in the corneal stroma without causing signs of edema such that it is equal to the SP (described as a mean level of approximately 55 mmHg); imbibition pressure (IP), which is related to endothelial barrier function and represents the negative pressure that draws fluid into the cornea in healthy individuals; and intraocular pressure (IOP), which represents the aqueous humor pressure against the corneal endothelium. All these corneal pressure equivalents are related to each other and can be described by the following equation:

When IP ≤ 0, there is no corneal edema because IOP does not exceed the swelling pressure (SP) level. When IP > 0, corneal edema appears because IOP > SP (i.e., the IOP >≈ 55 mmHg) (Figure 7)35,36,37.

Schematic representation of IOP-dependent corneal swelling, which was used in the modified “Bead Method” as an indicator of successful injection.
Under normal conditions (A), when the IOP does not exceed the level of stromal swelling pressure (SP), negative imbibition pressure (IP), which is dependent upon an intact endothelial barrier, prevents humor movement toward the corneal stroma and the subepithelial space. When IOP increases (B) and exceeds the level of SP, the corneal endothelial barrier is disturbed, resulting in water accumulation in the stroma and subepithelial layers (corneal edema).
In our glaucoma model, the beads were injected such that corneal edema was obtained with sub-epithelial cystic changes within the first few minutes of the procedure. Based on the above relationships, the IOP attained in the eye during bead injection exceeded the SP level (≈55 mmHg), which mimicked an acute glaucoma attack. Under such conditions, reliable tonometric measurements were impossible because of the presence of the glass needle in the anterior chamber. The edema disappeared within the first 6 hours, after which full corneal transparency was restored.
Intraocular pressure
IOP was measured with a rebound TonoLab tonometer at 1 and 7 days after injection of the beads and then once per week for 6 weeks until the animals were euthanized. No anesthesia was used during the IOP measurements. IOP measurements were always carried out at the same time of the day; three measurements were performed each time and the mean values were calculated.
Fundus photographs
Fundus photographs were captured to examine optic nerve disc parameters. Only animals with good optical center transparency were included in the study (i.e., eyes in which the beads had spread into the iridocorneal angle, without cataract or corneal ulceration (n = 10 glaucomatous eyes, n = 10 healthy eyes)). Photographs were taken 6 weeks after injection of the beads. The procedure required systemic anesthesia and mydriasis (0.5% Tropicamide, Santen Oy, Helsinki, Finland). Fundus photographs were captured with a fundus camera (Topcon TRC-NW7 SF, Topcon, Japan) combined with a +20.0 D Volk lens (Volk, Mentor, OH, US). In the obtained photographs, cup/disc (c/d) ratio changes were analyzed.
Immunohistochemistry methods
Five animals were sacrificed after 1 week to exclude signs of early, severe retinal damage (due to ischemia). Other animals were sacrificed after 6 weeks as following. The animals were anesthetized and perfused transcardially for 5 minutes with 0.1 M PBS followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 10 min. The eyeballs, with retro-bulbar stumps of optic nerves, were removed and post-fixed in 4% paraformaldehyde for 3 hours.
Optic nerve processing
After fixation, the optic nerves were dissected from the eyeballs. Five out of 20 optic nerves from each group (glaucoma and healthy) were embedded in optimal cutting temperature compound (Tissue-Tek® O.C.T.™ Compound, Sakura, Netherlands) and sectioned at a thickness of 10 μm in a Thermo Scientific cryostat (Thermo Scientific, Waltham, MA, US). The sections were then mounted onto glass slides coated with poly-L-lysine (Sigma, St. Louis, MO, USA) and processed for immunostaining. Other optic nerve stumps were incubated overnight at room temperature in 1% osmium oxide (VIII) saline solution, washed in PBS, dehydrated in ascending ethanol solutions and embedded in epoxy resin (Sigma-Aldrich, St. Louis, MO, US). The cross-sections (1 μm) of the optic nerves were stained with 1% toluidine blue and used for counting axons.
Processing of the retinas
Retinas from five eyeballs in each group (glaucoma and healthy) were removed and processed for immunostaining as “whole mounted” retinas. Other eyeballs were embedded in paraffin, sectioned at a thickness of 5 μm using a Leica Jung RM2035 microtome (Leica, Germany), collected on slides, deparaffinized and hydrated and processed for immunostaining to assess the number of RGCs.
Tissue staining
To visualize beads in the anterior chamber and the iridocorneal angle, eyeball cross-sections were stained with Harris's hematoxylin and eosin (Sigma-Aldrich, St. Louis, MO, US).
To visualize apoptotic cells, we performed TUNEL staining of paraffin sections. Briefly, the sections were incubated with 0.1 M Tris-HCl (pH = 7.5)/3% BSA/20% NGS for 30 minutes followed by a 1-hour incubation with the In Situ Cell Death Detection Kit at 37°C (Roche, Mannheim, Germany). The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, St. Louis, MO, USA) fluorescent stain (1:10,000 in 0.05 M Tris buffered saline) for 10 minutes and mounted with Mowiol mounting medium (Calbiochem, CA, USA).
Cryosections of optic nerves as well as paraffin sections of whole mounted retinas and eyeballs were incubated with a blocking solution containing 20% NGS/0.05 M TBS/0.5% Triton for 45 minutes to eliminate non-specific binding. Subsequently, the sections were incubated with primary antibody at +4°C overnight and with secondary antibody at RT for 3 hours. The samples were then washed twice in TBS and incubated for 15 minutes with DAPI (1:10,000, Sigma, St. Louis, MO, USA). The primary antibodies used were rabbit anti-β3-tubulin antibody (1:300, Cell Signaling Technology, Danvers, MA, US), which is a marker for retinal ganglion cells, mouse anti-NeuN antibody (1:500, Merck Millipore, Billerica, MA, US), which stains the nuclei of retinal neurons and rabbit anti-GFAP antibody (1:500, Merck Millipore, Billerica, MA, US), which is used to label glial cells. The secondary antibodies–goat anti-rabbit IgG AlexaFluor 488/594 and goat anti-mouse IgG AlexaFluor 488/594 (both 1:500, Life Technologies, Carlsbad, CA, USA)–were related to the applied primary antibodies. The samples were examined under a fluorescence microscope (Nikon, Japan).
RGCs and optic nerve axon counting
Cell survival was evaluated by counting the number of cells that were positively stained for DAPI (nuclei of all retinal cells), β3-tubulin (RGC bodies) and NeuN (retinal neuron nuclei) within the ganglion cell layer in 10 corresponding serial cross-sections throughout the eye (from healthy and glaucoma group), as previously described by Lorber B et al.38,39 Counting was performed using a fluorescence microscope (Nikon, Japan) with 20× magnification. After the length of each retinal section was measured (ImageJ, NIH, USA), the cells were counted in the whole section length (from ora serrata to ora serrata) and the number of cells was recalculated per 200 μm of retinal section, due to different sections length. Finally, the mean number of cells per each eye was calculated.
Fixed-pattern sampling methods described previously were used for counting axons40. The number of axons was estimated according to section area and presented as total axon number and axon density per μm2.
Statistical analysis
Statistical analysis was performed with IBM SPSS STATISTICS 20 (IBM, Armonk, NY, USA). Descriptive statistical results were presented using the mean values (mean) ± standard deviation (SD). The Kolmogorov–Smirnov test was used to determine whether the data were normally distributed. Comparisons between groups were performed using a U-Mann Whitney or paired samples Wilcoxon test. A P value < 0.05 was considered statistically significant.
References
Quigley, H. A. Glaucoma. Lancet. 377, 1367–1377 (2011).
Flammer, J. & Mozaffarieh, M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv. Ophthalmol. 52, 162–173 (2007).
Nickells, R. W. From ocular hypertension to ganglion cell death: a theorical sequence of events leading to glaucoma. Can. J. Ophthalmol. 42, 278–287 (2007).
Flammer, J. et al. The eye and the heart. Eur. Heart. J. 34, 1270–1278 (2013).
Gugleta, K. et al. Association between risk factors and glaucomatous damage in untreated primary open-angle glaucoma. J. Glaucoma. 22, 501–505 (2013).
Mozaffarieh, M. & Flammer, J. New insights in the pathogenesis and treatment of normal tension glaucoma. Curr. Opin. Pharmacol. 13, 43–49 (2013).
Kass, M. A. et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch. Ophthalmol. 120, 701–713, discussion 829–830 (2002).
Leske, M. C. et al. Factors for glaucoma progression and the effect of treatment: the early manifest glaucoma trial. Arch. Ophthalmol. 121, 48–56 (2003).
Musch, D. C., Gillespie, B. W., Lichter, P. R., Niziol, L. M. & Janz, N. K. Visual field progression in the Collaborative Initial Glaucoma Treatment Study the impact of treatment and other baseline factors. Ophthalmology. 116, 200–207 (2009).
Cone, F. E., Gelman, S. E., Son, J. L., Pease, M. E. & Quigley, H. A. Differential susceptibility to experimental glaucoma among 3 mouse strains using bead and viscoelastic injection. Exp. Eye Res. 91, 415–424 (2010).
McKinnon, S. J., Schlamp, C. L. & Nickells, R. W. Mouse models of retinal ganglion cell death and glaucoma. Exp. Eye Res. 88, 816–824 (2009).
Vecino, E. & Sansar, C. S. Glaucoma Animal Models., in: Rumelt, S. (Eds.), Glaucoma - Basic and Clinical Concepts. InTech, Rijeka, 319–334 (2011).
Urcola, J. H., Hernández, M. & Vecino, E. Three experimental glaucoma models in rats: comparison of the effects of intraocular pressure elevation on retinal ganglion cell size and death. Exp. Eye Res. 83, 429–437 (2006).
Aung, T. et al. Changes in retinal nerve fiber layer thickness after acute primary angle closure. Ophthalmology. 111, 1475–1479 (2004).
Morrison, J. C., Johnson, E. & Cepurna, W. O. Rat models for glaucoma research. Prog. Brain Res. 173, 285–301 (2008).
Sharmini, A. T., Yin, N. Y., Lee, S. S., Jackson, A. L. & Stewart, W. C. Mean target intraocular pressure and progression rates in chronic angle-closure glaucoma. J. Ocul. Pharmacol. Ther. 25, 71–75 (2009).
Goldblum, D. & Mittag, T. Prospects for relevant glaucoma models with retinal ganglion cell damage in the rodent eye. Vision Res. 42, 471–478 (2002).
Pang, I. H. & Clark, A. F. Rodent models for glaucoma retinopathy and optic neuropathy. J. Glaucoma. 16, 483–505 (2007).
Pease, M. E., Cone, F. E., Gelman, S., Son, J. L. & Quigley, H. A. Calibration of the TonoLab tonometer in mice with spontaneous or experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 52, 858–864 (2011).
Cone, F. E. et al. The effects of anesthesia, mouse strain and age on intraocular pressure and an improved murine model of experimental glaucoma. Exp. Eye Res. 99, 27–35 (2012).
Frankfort, B. J. et al. Elevated intraocular pressure causes inner retinal dysfunction before cell loss in a mouse model of experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 54, 762–770 (2013).
Sappington, R. M., Carlson, B. J., Crish, S. D. & Calkins, D. J. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest. Ophthalmol. Vis. Sci. 51, 207–216 (2010).
Kalesnykas, G. et al. Retinal ganglion cell morphology after optic nerve crush and experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 53, 3847–3857 (2012).
Cone-Kimball, E. et al. Scleral structural alterations associated with chronic experimental intraocular pressure elevation in mice. Mol. Vis. 19, 2023–2039 (2013).
Samsel, P. A., Kisiswa, L., Erichsen, J. T., Cross, S. D. & Morgan, J. E. A novel method for the induction of experimental glaucoma using magnetic microspheres. Invest. Ophthalmol. Vis. Sci. 52, 1671–1675 (2011).
Dai, C. et al. Olfactory Ensheathing Cells Rescue Optic Nerve Fibers in a Rat Glaucoma Model. Transl. Vis. Sci. Technol. 1, 3 (2012).
Dai, C. et al. Structural basis of glaucoma: the fortified astrocytes of the optic nerve head are the target of raised intraocular pressure. Glia. 60, 13–28 (2012).
Jay, J. L. & Murdoch, J. R. The rate of visual field loss in untreated primary open angle glaucoma. Br. J. Ophthalmol. 77, 176–178 (1993).
Heijl, A., Bengtsson, B., Hyman, L., Leske, M. C. & Early Manifest Glaucoma Trial Group. Natural history of open-angle glaucoma. Ophthalmology. 116, 2271–2276 (2009).
Hyman, L., Heijl, A., Leske, M. C., Bengtsson, B., Yang, Z. & Early Manifest Glaucoma Trial Group. Natural history of intraocular pressure in the early manifest glaucoma trial: A 6-year follow-up. Arch. Ophthalmol. 128, 601–607 (2010).
Rossetti, L. et al. Focusing on glaucoma progression and the clinical importance of progression rate measurement: a review. Eye (Lond). 24, S1–S7 (2010).
Peters, D., Bengtsson, B. & Heijl, A. Lifetime risk of blindness in open-angle glaucoma. Am. J. Ophthalmol. 156, 724–730 (2013).
Heijl, A., Bengtsson, B. & Oskarsdottir, S. E. Prevalence and severity of undetected manifest glaucoma: results from the early manifest glaucoma trial screening. Ophthalmology. 120, 1541–1545 (2013).
Melamed, S., Ben-Sira, I. & Ben-Shaul, Y. Corneal endothelial changes under induced intraocular pressure elevation: a scanning and transmission electron microscopic study in rabbits. Br. J. Ophthalmol. 64, 164–169 (1980).
Ytteborg, J. & Dohlman, C. H. Corneal edema and ocular pressure. I. Animal experiments. Arch. Ophthalmol. 74, 375–381 (1965).
Ytteborg, J. & Dohlman, C. H. Corneal edema and intraocular pressure: II. Clinical results. Arch. Ophthalmol. 74, 477–484 (1965).
Bolívar, G., Teus, M. & Arranz-Marquez, E. Effect of acute increases of intraocular pressure on corneal pachymetry in eyes treated with travoprost: an animal study. Curr. Eye Res. 36, 1014–1019 (2011).
Lorber, B., Howe, M. L., Benowitz, L. I. & Irwin, N. Mst3b, an Ste20-like kinase, regulates axon regeneration in mature CNS and PNS pathways. Nat. Neurosci. 12, 1407–1414 (2009).
Lorber, B., Guidi, A., Fawcett, J. W. & Martin, K. R. Activated retinal glia mediated axon regeneration in experimental glaucoma. Neurobiol. Dis. 45, 243–252 (2012).
Ebneter, A., Casson, R. J., Wood, J. P. & Chidlow, G. Estimation of axon counts in a rat model of glaucoma: comparison of fixed-pattern sampling with targeted sampling. Clin. Experiment. Ophthalmol. 40, 626–633 (2012).
Hernandez, M., Rodriguez, F. D., Sharma, S. C. & Vecino, E. Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol. Vis. 15, 2696–709 (2009).
Author information
Authors and Affiliations
Contributions
S.A. and P.-D.M. designed the study, executed the experiments (both in vivo experiments and tissue analysis), interpreted the data, drafted the manuscript and created the figures. K.K. and L.-K.J. conceived the study, supervised the experiments and critically reviewed the manuscript. K.K. performed the statistical analysis. L.-K.J. performed the literature review.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Smedowski, A., Pietrucha-Dutczak, M., Kaarniranta, K. et al. A rat experimental model of glaucoma incorporating rapid-onset elevation of intraocular pressure. Sci Rep 4, 5910 (2014). https://doi.org/10.1038/srep05910
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05910
This article is cited by
-
Corneal confocal microscopic characteristics of acute angle-closure crisis
BMC Ophthalmology (2022)
-
Differential susceptibility of retinal ganglion cell subtypes in acute and chronic models of injury and disease
Scientific Reports (2020)
-
Candidate proteins from predegenerated nerve exert time-specific protection of retinal ganglion cells in glaucoma
Scientific Reports (2017)
-
Immune response after intermittent minimally invasive intraocular pressure elevations in an experimental animal model of glaucoma
Journal of Neuroinflammation (2016)
-
Predegenerated Schwann cells–a novel prospect for cell therapy for glaucoma: neuroprotection, neuroregeneration and neuroplasticity
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
