
Abstract
The α-amylases from Anoxybacillus species (ASKA and ADTA), Bacillus aquimaris (BaqA) and Geobacillus thermoleovorans (GTA, Pizzo and GtamyII) were proposed as a novel group of the α-amylase family GH13. An ASKA yielding a high percentage of maltose upon its reaction on starch was chosen as a model to study the residues responsible for the biochemical properties. Four residues from conserved sequence regions (CSRs) were thus selected and the mutants F113V (CSR-I), Y187F and L189I (CSR-II) and A161D (CSR-V) were characterised. Few changes in the optimum reaction temperature and pH were observed for all mutants. Whereas the Y187F (t1/2 43 h) and L189I (t1/2 36 h) mutants had a lower thermostability at 65°C than the native ASKA (t1/2 48 h), the mutants F113V and A161D exhibited an improved t1/2 of 51 h and 53 h, respectively. Among the mutants, only the A161D had a specific activity, kcat and kcat/Km higher (1.23-, 1.17- and 2.88-times, respectively) than the values determined for the ASKA. The replacement of the Ala-161 in the CSR-V with an aspartic acid also caused a significant reduction in the ratio of maltose formed. This finding suggests the Ala-161 may contribute to the high maltose production of the ASKA.
Similar content being viewed by others

Introduction
Most α-amylases (EC 3.2.1.1) belong to family 13 of glycoside hydrolases (GH13)1,2. The polyspecific family GH13 covering more than 30 different amylolytic and related enzyme specificities with nearly 16,000 sequences ranks among the largest of the GH families2. It is also the main α-amylase family of the α-amylase CAZy clan GH-H that is formed by the families GH13, GH70 and GH773,4,5,6,7. The family GH13 has been divided into curator-based subfamilies (currently 40)8, although some of them were previously established9,10 and others have not yet been established11. α-Amylase is a very common enzyme produced by most living organisms3. Because it is necessary for the digestion of complex starchy substrates in living systems12, it is also important in various branches of industrial production13,14. In addition to other bacterial producers, several Anoxybacillus species have been shown to produce α-amylases with potentially industry-desired properties including the ability to digest raw starch15,16,17,18,19.
Recently, we described the biochemical properties of two Anoxybacillus α-amylases produced by the closely related Anoxybacillus sp. SK3-4 (ASKA) and Anoxybacillus sp. DT3-1 (ADTA)18. Interestingly, although both these α-amylases belong to the main α-amylase family GH13, the phylogenetic tree clearly demonstrates that their cluster is independent of all existing GH13 subfamilies18. A few other α-amylases exhibiting close relatedness to ASKA and ADTA were reported, i.e., those from B. aquimaris11 and G. thermoleovorans20,21,22. This novel, currently undefined GH13 subfamily of α-amylases may be most closely related to subfamily GH13_1 of extracellular fungal α-amylases2. It, however, can be distinguished from other GH13 subfamilies with the α-amylase specificity by the exclusive presence of two consecutive tryptophan residues (Trp-201 and Trp-202; B. aquimaris α-amylase numbering) positioned at helix α3 of the catalytic TIM-barrel domain11. Of the three α-amylases from G. thermoleovorans, two of them are identical (amyA from the strain PizzoT and GTA from the strain CCB US3 UF5)21,22 and share 94% sequence identity with the third one (Gt-amyII from the strain MTCC 4220)20.
A preliminary inspection of the CSRs revealed that several positions are not conserved in ASKA when compared with α-amylase counterparts from other GH13 subfamilies. The main goal of the present study was to determine the function of some distinct amino acid residues in the ASKA CSRs using site-directed mutagenesis. These residues including Phe-113 in CSR-I, Tyr-187 and Leu-189 in CSR-II and Ala-161 in CSR-V (ASKA numbering) were substituted with valine, phenylalanine, isoleucine and aspartic acid, respectively, which are more conserved in other GH13 subfamilies containing the α-amylase specificity.
Results
Analysis of the conserved sequence regions of α-amylases
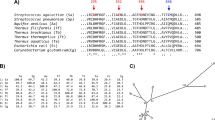
ASKA, ADTA, Pizzo, GtamyII, GTA and BaqA α-amylases phylogenetically cluster together, creating a new GH13 subfamily. Other homologs of this subfamily are summarised in Table 1, based on the BLAST results (see the Methods section). Most of these α-amylases are not well characterised. Using the thirty-six α-amylases shown in Table 1, seven conserved sequence regions (CSRs) typical for the members of this new α-amylase family GH1323 were identified in these sequences and the weblogo covering the CSRs was created (Figure 1). Compared with the representatives of this new GH13 subfamily, ASKA and all its closely homologous subfamily counterparts have similar CSRs (Figure 1). A separate weblogo with α-amylase sequences from earlier known GH13 subfamilies was built and compare with ASKA's CSRs (Figure 2). ASKA clearly contains different residues in several positions of the CSRs (Figure 2). To investigate their functional role, Phe-113, Ala-161, Tyr-187 and Leu-189 of ASKA were thus selected and replaced with valine, aspartic acid, phenylalanine and isoleucine, respectively. A total of four single-point mutant α-amylases were designed and denoted as F113V, A161D, Y187F and L189I (Table 2).

Sequence logo for the seven CSRs of the GH13 subfamily represented by ASKA.
CSR-I, residues 10–15; CSR-II, residues 23–31; CSR-III, residues 32–39; CSR-IV, residues 40–45; CSR-V, residues 16–20; CSR-VI, residues 1–9; and CSR-VII, residues 46–54. The two characteristic, consecutive tryptophans are also shown. The catalytic triad of the GH13 family, i.e., the catalytic nucleophile (No. 27, aspartic acid), the proton donor (No. 36, glutamic acid) and the transition-state stabiliser (No. 45, aspartic acid) are indicated with asterisks. The logo is based on the sequences of 36 proteins including both the characterised α-amylases and very similar hypothetical proteins (Table 1). The corresponding ASKA CSR sequences are stated above the logo and the mutated residues described in this study are shown in red.

Sequence logos of the seven CSRs based on the mixture of various GH13 subfamily α-amylases (Table S1).
The size of the single letter amino acid code in each sequence logo represents the occurrence of a particular amino acid at a particular position. The corresponding amino acid sequences of ASKA were stated above each sequence logo and the mutated residues performed in this study were shown in red.
Figure 3a illustrates the position of CSR-I to CSR-VII in silico. Phe-113 is the second residue of CSR-I (Figure 1; position 11 in the logo), which is located on strand β3 of the catalytic (β/α)8-barrel domain A23. This position was chosen for the mutagenesis study because a valine or other aliphatic hydrophobic residue has frequently been observed at the corresponding position, e.g., in the α-amylases from subfamilies GH13_1 (fungi), GH13_5 (liquefying enzymes from bacteria), GH13_6 (plants) and GH13_7 (archaeons)23,24. The next two mutant positions, Tyr-187 and Leu-189 (logo positions 24 and 26, respectively; Figure 1), are residues from CSR-II, which covers strand β4 of the catalytic barrel23. While the former position is well-conserved as an aromatic one throughout the entire α-amylase family GH13, it is mostly occupied by phenylalanine or tryptophan23,24 and the presence of the tyrosine (Figure 2) here in ASKA seems to be unique for the GH13 subfamily11. Because tryptophan is highly characteristic of the GH13 subfamilies 6 and 7, which include plants, archaeons and flavobacteria23,24,25, phenylalanine, which is found in the GH13 subfamilies 15 and 24 (animals) as well as 27, 28, 32 and 36 (various groups of bacteria), was chosen as the substitute for Tyr-187 in the mutant. The latter mutant position in CSR-II, Leu-189, is a conserved position, but if it is replaced then isoleucine (Figure 2) is often found23,24. The fourth residue selected for mutation, Ala-161, is positioned in CSR-V near the C terminal of domain B, i.e., in the loop connecting stand β3 to helix α3 of catalytic (β/α)8-barrel domain A23. Although various residues can be seen at that position, aspartic acid or asparagine may represent a consensus23,24. It should therefore be of interest to investigate the role the alanine may play in ASKA (Figure 1; position 20 in the logo) because an alanine is typically present in the corresponding position only in the animal α-amylases (from vertebrates) from the subfamily GH13_2423.

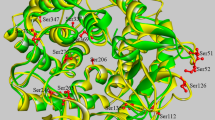
(a). Structural prediction of native ASKA. Green sphere, Calcium; grey ligand, substrate; yellow, three catalytic residues D190, E219 and D287; blue violet, CSR-1; violet, CSR-II; apple green, CSR-III; maroon, CSR-IV; red, CSR-V; forest green, CSR-VII; blue, CSR-VII. (b). The mutation sites F113, Y187 and L189 are located adjacent to the catalytic residues. (c). Structure prediction for mutant A161D. The putative residues for subsites −1 and +1 are shown in yellow and the side chain of mutant A161D is shown in red. The average distance from the A161D side chain to catalytic residue D190 is 18 Å.
Characterisation of the mutant α-amylases
The purified enzymes were subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) to determine the purity of the proteins (Figure 4). The mutant enzymes were pure with single bands at the molecular mass of 50 kDa in agreement with that of native α-amylase18. The zymogram analysis was performed using native PAGE and the gel was stained with iodine solution. For all the enzymes, a single, clear band was observed upon staining. The zymogram of the purified proteins shows that the mutant enzymes retained amylolytic activity (Figure 4a). After the purification procedures, the enzyme activity and the protein concentration for all α-amylase variants were determined and the specific activity of each α-amylase variant was calculated (Table 2).

(a). Zymogram analysis of the purified native and mutant α-amylases. (b). SDS-PAGE. Lane M: Promega™ Broad Range protein marker, Lane 1: Native ASKA, Lane 2: Mutant F113V, Lane 3: Mutant A161D, Lane 4: Mutant Y187F, Lane 5: Mutant L189I.
The effect of temperature on the activity of the ASKA and mutant variants was tested at temperatures ranging from 30 to 90°C at pH 8.0 (Supplemental file). The optimum reaction temperature for all the α-amylase variants was 60°C. None of the amino acid substitutions affected the optimum temperature activity. At 70°C, the enzymes maintained 54–87% of the initial activity. As the incubation temperature rises, the activity of the enzymes drops to 13–24% at 80°C and 10–18% at 90°C. Compared to ASKA, mutants Y187F and L189I had a slight decline in activity of approximately 32% and 23%, respectively at a temperature of 70°C. The thermostability of the enzymes was studied at the temperatures of 60°C and 65°C for 72 h. The activity half-life of all mutants at their optimum temperature of 60°C is unaffected by the mutation (Supplemental file). The mutant A161D retained 94.6% of its total activity after 72 h of incubation at 60°C and this value is slightly higher compared to ASKA, which retained 91% of its activity. The activity half-life at 65°C is displayed in Table 2, the ASKA had a half-life of 48 h, while Y187F and L189I had a slight reduction in thermostability with half-lives of 43 and 36 h, respectively. The activity half-lives at 65°C for the mutants F113V and A161D were 51 and 53 h, respectively.
The effect of pH on α-amylase activity and stability were tested at a pH range of 4.0 to 11.0 using sodium acetate buffer (pH 4–5), sodium phosphate buffer (pH 6–7), Tris-HCl buffer (pH 8–9) and glycine-NaOH buffer (pH 10–11) at a temperature of 60°C. ASKA and all mutant α-amylases exhibited maximum activity at pH 8.0. The optimum pH is thus unaffected by the amino acid substitutions. This result is expected because very frequently the optimum pH of an enzyme is not easily affected by a single residue replacement. The ASKA and mutant α-amylases were stable within a wide pH range of 7.0 to 10.0 for 30 min and the enzyme activities were at least 90% of the initial enzyme activity at these pH values. The pH stability of the mutants was similar to the ASKA. The pH stability of ASKA was unaffected by the mutations.
The specific activities, turnover rate (kcat) and catalytic efficiency (kcat/Km) of ASKA were compared to its mutant variants (Table 2). ASKA had a specific activity of 34 U/mg-protein. Of the mutant α-amylases, the mutant A161D had the highest specific activity at 76 U/mg with an increase of approximately 1.23 times. The Vmax as well as the kcat/Km were drastically changed by the A161 mutation. The mutant A161D exhibited the highest kcat of 39,108 s−1, which is 1.17-times higher than the kcat value of ASKA. Moreover, the kcat/Km of A161D was also higher than that of ASKA by 2.88-times, at 57,463 ml mg−1 s−1. Of the four mutants constructed using site-directed mutagenesis, only mutant A161D exhibited enhanced kcat and kcat/Km values (Table 2). The L189I substitution had an undesirable effect as the kcat and kcat/Km values were reduced by 81% and 74%, respectively. Such deterioration was the worst observed of the mutants. The adjacent mutant Y187F also exhibited a reduced efficiency in the catalytic reaction. Both of these residues were located near the catalytic Asp-190 (Figure 3b) and their mutations have caused a detrimental effect to the catalytic property of the enzyme. These results are in agreement with the findings of earlier reports26,27, which stated that any mutations near the catalytic residue will most likely influence the catalytic property of enzyme and likely cause an overall negative effect on enzyme catalysis.
All mutant enzymes were subjected to an enzymatic reaction to study the effect of the mutation on product specificity. Hydrolysis of 1% (w/v) and 4% (w/v) gelatinised soluble-, tapioca-, potato-, corn- and sago-starch was carried out at 60°C for 24 h. The total sugar production (mg/ml) is shown in Table 3. At the lower starch concentration of 1%, the yield of total sugars generated by mutants Y187F and L189I was significantly lower than that of the ASKA; however, this effect was less significant at the higher concentration of starch (4%). With the 1% starch solution, the total sugars yielded by mutant A161D were quite similar to the ASKA most likely because the amount of substrate was not in excess. Interestingly, at a higher substrate concentration, for instance 4% corn-starch, the total sugar formed by mutant A161D was 27.8 mg/ml, which is higher than that of the ASKA (16.3 mg/ml) (Table 3). The kcat and kcat/Km values for mutant A161D are better than for ASKA and the HPLC analysis confirms the outperformance of A161D.
The percentages of individual sugars that were formed after 24-h reactions in 1% (Figure 5a) and 4% corn-starch (Figure 5b) were studied. Previously, we reported that ASKA is a maltose-forming α-amylase18 and a negligible percentage of glucose (G1), maltohexaose (G6) and maltoheptaose (G7) were usually detected in 1% gelatinised corn-starch (Figure 5a). However, when the ASKA was reacted with 4% corn-starch for 24 h, a small percentage of G6 and G7 were detected. The product specificity spectrum for mutant A161D changed significantly for the reaction that was carried out in 4% starch. The percentage of maltose forming dropped, while a substantial increase in the percentage of glucose was observed (Figure 5b).

End product distribution of ASKA and mutant α-amylases from the hydrolysis of a) 1% (w/v) and b) 4% (w/v) corn-starch.
The data are shown as the percentage (%) hydrolysed. Black: glucose (G1), grey: maltose (G2), blue: maltotriose (G3), green: maltotetraose, (G4), purple: maltopentaose (G5), yellow: maltohexaose (G6) and red: maltoheptaose (G7).
Discussion
α-Amylases, in addition to all members of the α-amylase GH13 family, contain four to seven conserved sequence regions (CSRs), roughly covering strands β2, β3, β4, β5, β7 and β8 of the catalytic TIM-barrel and a short stretch near the C-terminus of domain B protruding out of the barrel in the β3-α3 loop. The individual α-amylases often possess unique sequence features in their respective CSRs that distinguish them from each other and thus these regions may be connected to the specific biochemical properties of each protein, such as substrate preferences, product specificities and thermostability9,10,11.
Anoxybacillus sp. SK3-4 was isolated from a Malaysian hotspring28 and its genome was sequenced using Illumina MiSeq29. Most of the glycoside hydrolases of the cell are expressed intracellularly; however, ASKA was described as an extracellular hydrolase18. Compared with well-recognised α-amylases, the ASKA protein sequence has a low similarity of 49% to the G. stearothermophilus30 (BSTA, subfamily GH13_5, UniProt Accession number P06279), 47% to the Aspergillus oryzae31 (TAKA, GH13_1, P0C1B3) and 37% similarity to the B. licheniformis32 (BLA, GH13_5, P06278) α-amylases. Despite the fact that ASKA is a high maltose producing α-amylase, it is a genuine α-amylase. In fact, the two types of family GH13 amylases forming mainly maltose (EC 3.2.1.133) should be distinguished from each other, i.e., maltogenic amylases and maltogenic α-amylases23. Whereas the former enzymes are members of the so-called neopullulanase subfamily (subfamily GH13_20), the latter ones are closely related to cyclodextrin glucanotransferases (CGTase) (GH13_2). A typical GH13_20 maltogenic amylase possesses in its CSR-V the signature MPKLN9, contains at its N-terminus a carbohydrate-binding module from the family CBM3433 and is able to hydrolyse cyclodextrins34. Because the ASKA has a different CSR-V (157_LPDLA; Figure 1), lacks the CBM34 N-terminal domain and is not active on cyclodextrins18, it is likely not related to this type of maltogenic amylase. For the maltogenic α-amylases from the subfamily GH13_2, the CSR-V signature (LADLS) from B. stearothermophilus23 is more similar to that seen in ASKA, but it typically shares its five-domain organisation with CGTase35; i.e., it has two C-terminal domains in addition to domains A, B and C that are present in the ASKA. Thus, ASKA and most likely the entire newly established α-amylase family GH13 group11 (i.e., ADTA, BaqA and homologues from G. thermoleovorans) may represent a novel type of high maltose producing amylase.
ASKA shares 69% similarity with the α-amylase from G. thermoleovorans subsp. stromboliensis Pizzo22, 69% to G. thermoleovorans GtamyII20 and 60% to a marine B. aquimaris MKSC 6.2 α-amylase (BaqA)11,36. Recently, the crystal structure of the α-amylase G. thermoleovarans (GTA) was reported21. The sequence similarity between ASKA and GTA is as high as 69%. ASKA, Pizzo, GtamyII, GTA and BaqA as well as another Anoxybacillus α-amylase ADTA from our laboratory18 can be classified as a new sub-group of the α-amylase family GH13. Taken together, the reported experimental data of these α-amylases, this new subgroup has at least three unique characteristics. Firstly, they are high maltose producing enzymes as seen in ASKA, ADTA and Pizzo and potential in GTA since Pizzo shares an identical protein sequence with GTA21. Secondly, these enzymes are able to degrade raw starches as reported for the Pizzo, GtamyII and BaqA amylases11,20,22. Although the two consecutive tryptophans were proposed to be the putative raw starch binding residues11, the binding action has yet to be experimentally proven. Thirdly, the C-terminal of the ASKA protein sequence was predicted to be a putative transmembrane region that could possibly anchor to the wild type Anoxybacillus sp. SK3-419. Recent work on the deletion of this transmembrane region of GtamyII elucidated its role in raw starch adsorption20.
The main goal of this current work is to address the reason that the enzymes from this new GH13 subgroup, for instance ASKA, are a high maltose forming. The CSRs of ASKA were compared to other reported α-amylases and four single point mutations were carried out. Interestingly, the product specificity of mutant A161D was altered causing the overall ratio of maltose to drop, while the percentage of glucose increased 28-times in 4% corn-starch. Based on these findings, Ala-161 is proposed to be responsible for the high maltose-producing characteristic of ASKA. In the maltose producing ADTA, Pizzo and GTA α-amylases, Ala is well conserved at an identical position. The other homologs, i.e., GtamyII and BaqA also contained Ala, but their product specificity has yet to be determined.
Such drastic change was unexpected because the location of 161D in the predicted 3D structure is 18 Å from the nearest catalytic residue, Asp-190 (Figure 3c). Moreover, the superimposition of the mutant A161D model with the G. thermoleovorans α-amylase (4E2O) elucidated that 161D has no direct interaction with the pseudo substrate acarbose. At this stage, it is not clear the true reason why product specificity is associated with Ala-161. In CGTase, the product specificity is often related to the type of amino acids at the subsite interacting residues37,38,39, yet A161 is not likely to be a part of any subsites. The in silico position of residue 161D is located close to the putative calcium (Ca2+)-binding site (Figure 3c). A161D may stabilise the secondary structure or the folding of this Ca2+-binding pocket, therefore the half-life of this mutant was higher than that of ASKA (Table 2). Nevertheless, at this point, we are unable to determine why A161D can alter the product specificity in a drastic manner. We are now conducting further examination of this site by creating other mutations to examine the effects of various amino acid side chains on product specificity. Besides X-ray crystallisation will be performed in the near future. We hope that these experiments will lead to more insight to the importance of A161 in product specificity.
Several mutations have previously been made to the CSRs of other α-amylases. For instance, Chen et al. (2010) mutated Asp-236 of the Bacillus sp. strain TS-23 α-amylase40. This mutated residue corresponds to Ala-161 in ASKA numbering, in CSR-V. The replacement of Asp-236 with asparagine resulted in a 33% reduction in the specific activity of the enzyme. Interestingly, this finding contradicts the A161D mutation results obtained in this study. In addition, Chen et al. (2010) mutated Asp-234, which is also located in CSR-V. This substitution resulted in an 86% reduction in the specific activity of the enzyme. It seemed from that study that two residues in CSR-V (Asp-234 and Asp-236) played a significant role in the catalytic properties of the enzyme40. Unfortunately, both trials disrupted the specific activity and the authors suggested that Asp-234 and Asp-236 were involved in Ca2+-binding and any changes to these Ca2+-binding residues will affect the enzyme's structural integrity and stability as well as its α-amylase activity. In another study, the substitution of Asp-233 of the B. amyloliquefaciens α-amylase (corresponds to ASKA's Ala-161) significantly affected the specific activity and thermostability of the enzyme. There was an 84.4% reduction in specific activity compared to the ASKA. From this result, the authors deduced that Asp-233 is important for the catalytic activity of the enzyme41.
Despite mutant F113V exhibiting a similar specific activity to ASKA, the kcat value for the mutant was 35% higher. ASKA and its homologues ADTA, Pizzo, GtamyII, BaqA and GTA have a Phe at this particular position of CSR-I, while most other α-amylases have another type of amino acid (Figure 2). Several mutations of CSR-I residues were performed previously, for instance, H137L (His-117 in ASKA) of the Bacillus sp. TS-23 α-amylase resulted in a complete loss of amylolytic activity42. Chang et al. (2003) explained that His-137 could be the essential catalytic residue in the amylolytic reaction. The site directed mutagenesis of another CSR-I residue, Asn-104 of BLA (corresponds to ASKA's Asn-116) was replaced with Asp. This mutant had an 11% decrease in specific activity at 30°C. But, interestingly, the mutant had a 30% higher specific activity than the ASKA at the higher temperature of 70°C43.
This study found that the mutation of CSR-II, i.e., mutant Y187F and L189I caused a more than 70% reduction in the kcat and kcat/Km (Table 2). For mutant Y187F, the chemical structures of tyrosine and phenylalanine are structurally close except that phenylalanine lacks a hydroxyl -OH group at end of the aromatic side chain. The removal of the hydroxyl group caused a severe disturbance during the starch hydrolysis process. The hydroxyl group is likely involved in stabilising of the intermediate state during the catalytic mechanism. These results elucidate that the amino acid residues Tyr-187 and Leu-189 are associated with ASKA activity, most likely because they are located adjacent to catalytic residue 190 (Figure 3b). Similarly, the mutation of the Bacillus sp. TS-23 α-amylase, in CSR-II at His-269 (TS-23 numbering), which is adjacent to catalytic Asp-265, led to the complete loss of amylolytic activity. The report proposed that this conserved histidine may play a substrate binding role37, or possibly, this stretch of CSR-II is sensitive to alteration.
Methods
Bioinformatics analysis
The 36 amino acid sequences (Table 1) that are currently thought to form the GH13 subfamily represented by ASKA were retrieved from GenBank database44. The sequences were collected from a BLAST search45 using as the query the stretch of 89 residues from the ASKA sequence18 spanning the polypeptide chain covering CSR-I (strand β3) and CSR-II (strand β4), including the entire domain B (GenBank Acc. No.: AFI49455.1: Asp-129-His-217). A sequence was considered to belong to this GH13 subfamily if it contains the features ascribed to the closely related α-amylase from B. aquimaris11, especially the two consecutive tryptophan residues in helix α3 of the catalytic TIM-barrel and the specific sequence in CSR-V positioned in domain B.
The sequences were aligned using the program Clustal-Omega46 available on the European Bioinformatics Institute's server (http://www.ebi.ac.uk/). For the whole set of 36 sequences, a sequence logo for their seven CSRs typical of the members of the α-amylase family GH1323 was created using the WebLogo 3.0 server47 (http://weblogo.berkeley.edu/).
Preparation of starting structure and molecular dynamics simulation
The protein structure of ASKA was constructed using the SWISS-MODEL48 program (http://swissmodel.expasy.org/) and the X-ray crystal structure of G. thermoleovorans α-amylase (PDB code: 4E2O)21 was selected as the template. Refinements of the model were performed using energy minimisation and dynamic simulation (MD). Prior to the MD simulation using the GROMACS software package49, calcium and glucose were removed from the structure. The system was solvated with SPC/E water in a dodecahedral box with 10 Å between the protein structure and the box. The system was subsequently neutralised by the addition of sodium ions. The MD simulation was performed using an OPLS-AA force field. The system was energy minimised and subsequently simulated in NVT and NPT ensembles. The temperature was maintained at 333 K. Upon completing the system equilibration, a production run of 10 ns was performed.
Mutagenesis
The α-amylase mutant gene was generated using the Overlap Extension PCR (OE-PCR) method. The external forward primer contained an EcoRI restriction site and the external reverse primer contained an XhoI restriction site. The change in nucleotide sequence was introduced by incorporating nucleotide changes into the overlapping oligo primers, as shown in Supplemental file. During the cloning process, the native signal peptide DNA sequence was excluded, as the pET-22b(+) contains a pelB leading sequence. Phusion® PCR Master Mix (NEB, Ipswich, Massachusetts, USA) was used to perform the PCR. PCR reaction mixtures containing 25 ng ASKA DNA template, 25 μl 2× PhusionTM Flash PCR Master Mix and 0.5 μM each primer were added to a final volume of 50 μl. A SpinPrepTM PCR Clean-Up kit (Novagen, Darmstadt, Germany) was used to purify the PCR products. Purified PCR products and pET-22b (+) (Novagen, Darmstadt, Germany) were digested with EcoRI and XhoI (NEB, Ipswich, Massachusetts, USA) and subsequently purified. The ligation was performed using T4 DNA ligase (NEB, Ipswich, Massachusetts, USA) and then transformed into E. coli DH5a. The recombinant plasmid was confirmed by sequencing. The recombinant plasmid was then transformed into the expression host E. coli BL21 (DE3) for later analysis.
Protein expression and purification
The recombinant E. coli BL21 (DE3) was cultured overnight in LB medium supplemented with 100 μg/ml ampicillin at 37°C with agitation at 200 rpm. Induction was performed by adding 0.01 mM IPTG and the cultivation was continued at 25°C with agitation at 200 rpm for 48 h50. The cell free enzyme was harvested after centrifugation (10000 × g, 20 min and 4°C). The crude enzyme solution was concentrated using the Vivaflow 50 crossflow system from Sartorius Stedim Biotech (Göttingen, Germany) using the polyethersulfone (PES) membrane with a 10 kDa molecular weight cut-off (MWCO). The retentate was loaded onto affinity columns and chromatography was conducted using the ÄKTAprime plus system (GE Healthcare, Uppsala, Sweden) according to the method reported previously18.
Characterisation of ASKA and mutant enzymes
The protocols used to characterise the mutant α-amylases were also performed using ASKA unless otherwise stated18. One unit of α-amylase was defined as the amount of enzyme that releases 1 μmol maltose per min per ml under the assay conditions specified. The kinetic parameters of the ASKA and mutant α-amylases were determined by measuring the amount of maltose production using the standard assay conditions. The samples were reacted with soluble starch (Kanto Chemical, Tokyo, Japan) at concentrations from 0.1 to 2.0 mg/ml in 0.1 M Tris-HCl buffer (pH 8.0). SigmaPlot v11.0 was used to build the Hanes plot.
The products of starch hydrolysis by α-amylase were analysed using High-performance liquid chromatography (HPLC). First, 0.1 ml purified enzyme was incubated with 1.0 ml 1% gelatinised starch in 0.1 M Tris-HCl buffer (pH 8.0) at 60°C for 24 h. The efficiency of degradation on various starches (soluble-, tapioca-, potato-, sago- and corn-starch) was compared. After 24 h of incubation, the reaction mixture was boiled for 15 min to stop the reaction and cooled to room temperature. The mixture was filtered through a 0.45 μm nylon syringe filter (Whatman, Little Chalfront, United Kingdom) to remove insoluble particles. The hydrolysis products were analysed using a Waters HPLC system with a Zorbax Carbohydrate analysis column, 4.6 × 250 mm (Agilent, Santa Clara, California, USA). An acetonitrile (HPLC grade, Merck, White House Station, New Jersey, USA) to water mixture (70:30, v/v) was used as the mobile phase and the flow rate was maintained at 1.0 ml/min. The column was kept at 30°C and a Waters 2414 refractive index detector was used to detect the reaction products. The data were recorded by the integrated computer system attached to the HPLC. Glucose, maltose, maltotriose, maltotetraose, maltopentaose, maltohexaose and maltoheptaose (Sigma-Aldrich. St Louis, Missouri, USA) were used as standards. Additionally, the efficiency of starch concentration on the hydrolysis products was investigated by incubating the ASKA and mutant enzymes in a 4% gelatinised starch in 0.1 M Tris-HCl buffer (pH 8.0) at 60°C for 24 h. The end products were analysed using HPLC. The assays and data collection were performed at least in triplicate, unless otherwise specified.
References
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238, 10.1093/nar/gkn663 (2009).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495, 10.1093/nar/gkt1178 (2014).
Janeček, Š., Svensson, B. & MacGregor, E. A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell. Mol. Life Sci. 71, 1149–1170, 10.1007/s00018-013-1388-z (2014).
Svensson, B. Protein engineering in the α-amylase family: catalytic mechanism, substrate specificity and stability. Plant Mol. Biol. 25, 141–157, 10.1007/bf00023233 (1994).
Janeček, Š. α-amylase family: molecular biology and evolution. Prog. Biophys. Mol. Biol. 67, 67–97, 10.1016/S0079-6107(97)00015-1 (1997).
Kuriki, T. & Imanaka, T. The concept of the α-amylase family: structural similarity and common catalytic mechanism. J. Biosci. Bioeng. 87, 557–565, 10.1016/S1389-1723(99)80114-5 (1999).
MacGregor, E. A., Janeček, Š. & Svensson, B. Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1546, 1–20, 10.1016/S0167-4838(00)00302-2 (2001).
Stam, M. R., Danchin, E. G. J., Rancurel, C., Coutinho, P. M. & Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562, 10.1093/protein/gzl044 (2006).
Oslancová, A. & Janeček, Š. Oligo-1,6-glucosidase and neopullulanase enzyme subfamilies from the α-amylase family defined by the fifth conserved sequence region. Cell. Mol. Life Sci. 59, 1945–1959, 10.1007/pl00012517 (2002).
Majzlová, K., Pukajová, Z. & Janeček, Š. Tracing the evolution of the α-amylase subfamily GH13_36 covering the amylolytic enzymes intermediate between oligo-1,6-glucosidases and neopullulanases. Carbohydr. Res. 367, 48–57, 10.1016/j.carres.2012.11.022 (2013).
Puspasari, F. et al. Raw starch-degrading α-amylase from Bacillus aquimaris MKSC 6.2: isolation and expression of the gene, bioinformatics and biochemical characterization of the recombinant enzyme. J. Appl. Microbiol. 114, 108–120, 10.1111/jam.12025 (2013).
Koropatkin, N. M., Cameron, E. A. & Martens, E. C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335, 10.1038/nrmicro2746 (2012).
Lévêque, E., Janeček, Š., Haye, B. & Belarbi, A. Thermophilic archaeal amylolytic enzymes. Enzyme Microb. Technol. 26, 3–14, 10.1016/S0141-0229(99)00142-8 (2000).
van der Maarel, M. J. E. C., van der Veen, B., Uitdehaag, J. C. M., Leemhuis, H. & Dijkhuizen, L. Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 94, 137–155, 10.1016/S0168-1656(01)00407-2 (2002).
Tawil, G., Viksø-Nielsen, A., Rolland-Sabaté, A., Colonna, P. & Buléon, A. Hydrolysis of concentrated raw starch: a new very efficient α-amylase from Anoxybacillus flavothermus. Carbohydr. Polym. 87, 46–52, 10.1016/j.carbpol.2011.07.005 (2012).
Matpan Bekler, F. & Güven, K. Isolation and production of thermostable α-amylase from thermophilic Anoxybacillus sp. KP1 from Diyadin hot spring in Ağri, Turkey. Biologia 69, 419–427, 10.2478/s11756-014-0343-2 (2014).
Agüloğlu Fincan, S., Enez, B., Özdemir, S. & Matpan Bekler, F. Purification and characterization of thermostable α-amylase from thermophilic Anoxybacillus flavithermus. Carbohydr. Polym. 102, 144–150, 10.1016/j.carbpol.2013.10.048 (2014).
Chai, Y. Y., Rahman, R. N. Z. R. A., Illias, R. M. & Goh, K. M. Cloning and characterization of two new thermostable and alkalitolerant α-amylases from the Anoxybacillus species that produce high levels of maltose. J. Ind. Microbiol. Biotechnol. 39, 731–741, 10.1007/s10295-011-1074-9 (2012).
Kahar, U., Chan, K.-G., Salleh, M., Hii, S. & Goh, K. M. A high molecular-mass Anoxybacillus sp. SK3-4 amylopullulanase: characterization and its relationship in carbohydrate utilization. Int. J. Mol. Sci. 14, 11302–11318, 10.3390/ijms140611302 (2013).
Mehta, D. & Satyanarayana, T. Domain C of thermostable α-amylase of Geobacillus thermoleovorans mediates raw starch adsorption. Appl. Microbiol. Biotechnol. 1–17, 10.1007/s00253-013-5459-8 (2014).
Mok, S.-C., Teh, A.-H., Saito, J. A., Najimudin, N. & Alam, M. Crystal structure of a compact α-amylase from Geobacillus thermoleovorans. Enzyme Microb. Technol. 53, 46–54, 10.1016/j.enzmictec.2013.03.009 (2013).
Finore, I. et al. Purification, biochemical characterization and gene sequencing of a thermostable raw starch digesting α-amylase from Geobacillus thermoleovorans subsp. stromboliensis subsp. nov. World J. Microbiol. Biotechnol. 27, 2425–2433, 10.1007/s11274-011-0715-5 (2011).
Janeček, Š. How many conserved sequence regions are there in the α-amylase family? Biologia - Section Cellular and Molecular Biology 57 (Suppl. 11), 29–41 (2002).
Janeček, Š., Lévêque, E., Belarbi, A. & Haye, B. Close evolutionary relatedness of α-amylases from Archaea and plants. J. Mol. Evol. 48, 421–426, 10.1007/PL00006486 (1999).
Li, C. et al. Close relationship of a novel Flavobacteriaceae α-amylase with archaeal α-amylases and good potentials for industrial applications. Biotechnol. Biofuels 7, 18, 10.1186/1754-6834-7-18 (2014).
Nielsen, J. E. et al. Electrostatics in the active site of an α-amylase. Eur. J. Biochem. 264, 816–824, 10.1046/j.1432-1327.1999.00664.x (1999).
Wind, R. D., Uitdehaag, J. C. M., Buitelaar, R. M., Dijkstra, B. W. & Dijkhuizen, L. Engineering of cyclodextrin product specificity and pH optima of the thermostable cyclodextrin glycosyltransferase from Thermoanaerobacterium thermosulfurigenes EM1. J. Biol. Chem. 273, 5771–5779 (1998).
Chai, Y. Y., Kahar, U. M., Salleh, M. M., Illias, R. M. & Goh, K. M. Isolation and characterization of pullulan-degrading Anoxybacillus species isolated from Malaysian hot springs. Environ. Technol. 33, 1231–1238, 10.1080/09593330.2011.618935 (2011).
Goh, K. M. et al. Analysis of Anoxybacillus genomes from the aspects of lifestyle adaptations, prophage diversity and carbohydrate metabolism. PLoS ONE 9, e90549, 10.1371/journal.pone.0090549 (2014).
Wind, R. D., Buitelaar, R. M., Eggink, G., Huizing, H. J. & Dijkhuizen, L. Characterization of a new Bacillus stearothermophilus isolate: a highly thermostable α-amylase-producing strain. Appl. Microbiol. Biotechnol. 41, 155-162, 10.1007/bf00186953 (1994).
Matsuura, Y., Kusunoki, M., Harada, W. & Kakudo, M. Structure and possible catalytic residues of Taka-amylase A. J. Biochem. 95, 697-702 (1984).
Ivanova, V. N., Dobreva, E. P. & Emanuilova, E. I. Purification and characterization of a thermostable α-amylase from Bacillus licheniformis. J. Biotechnol. 28, 277-289, 10.1016/0168-1656(93)90176-N (1993).
Kim, J.-S. et al. Crystal structure of a maltogenic amylase provides insights into a catalytic versatility. J. Biol. Chem. 274, 26279–26286 (1999).
Cha, H.-J. et al. Molecular and enzymatic characterization of a maltogenic amylase that hydrolyzes and transglycosylates acarbose. Eur. J. Biochem. 253, 251–262, 10.1046/j.1432-1327.1998.2530251.x (1998).
Dauter, Z. et al. X-ray structure of Novamyl, the five-domain “maltogenic” α-amylase from Bacillus stearothermophilus: maltose and acarbose complexes at 1.7A resolution. Biochemistry 38, 8385–8392, 10.1021/bi990256l (1999).
Puspasari, F. et al. Characteristics of raw starch degrading α-amylase from Bacillus aquimaris MKSC 6.2 associated with soft coral Sinularia sp. Starch - Stärke 63, 461–467, 10.1002/star.201000127 (2011).
Goh, K. M., Mahadi, N. M., Hassan, O., Abdul Rahman, R. N. Z. R. & Illias, R. M. The effects of reaction conditions on the production of γ-cyclodextrin from tapioca starch by using a novel recombinant engineered CGTase. J. Mol. Catal. B-Enzym. 49, 118–126, 10.1016/j.molcatb.2007.09.011 (2007).
Goh, K. M., Mahadi, N. M., Hassan, O., Rahman, R. N. Z. R. A. & Illias, R. M. A predominant β-CGTase G1 engineered to elucidate the relationship between protein structure and product specificity. J. Mol. Catal. B-Enzym. 57, 270–277, 10.1016/j.molcatb.2008.09.016 (2009).
Han, R. et al. Recent advances in discovery, heterologous expression and molecular engineering of cyclodextrin glycosyltransferase for versatile applications. Biotechnol. Adv. 32, 415–428, 10.1016/j.biotechadv.2013.12.004 (2014).
Chen, Y.-H. et al. Mutational analysis of the proposed calcium-binding aspartates of a truncated α-amylase from Bacillus sp. strain TS-23. Ann. Microbiol. 60, 307–315, 10.1007/s13213-010-0042-3 (2010).
Liu, Y., Shen, W., Shi, G.-Y. & Wang, Z.-X. Role of the calcium-binding residues Asp231, Asp233 and Asp438 in α-amylase of Bacillus amyloliquefaciens as revealed by mutational analysis. Curr. Microbiol. 60, 162–166, 10.1007/s00284-009-9517-5 (2010).
Chang, C.-T. et al. Identification of essential histidine residues in a recombinant α-amylase of thermophilic and alkaliphilic Bacillus sp. strain TS-23. Extremophiles 7, 505–509, 10.1007/s00792-003-0341-8 (2003).
Priyadharshini, R. & Gunasekaran, P. Site-directed mutagenesis of the calcium-binding site of α-amylase of Bacillus licheniformis. Biotechnol. Lett. 29, 1493–1499, 10.1007/s10529-007-9428-0 (2007).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 42, D32–D37, 10.1093/nar/gkt1030 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410, 10.1016/S0022-2836(05)80360-2 (1990).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539–544, 10.1038/msb.2011.75 (2011).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190, 10.1101/gr.849004 (2004).
Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201, 10.1093/bioinformatics/bti770 (2006).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854, 10.1093/bioinformatics/btt055 (2013).
Muniandy, K. et al. Application of statistical experimental design for optimization of novel α-amylase production by Anoxybacillus species. J. Biol. Sci. 13, 605–613, 10.3923/jbs.2013.605.613 (2013).
Acknowledgements
This study was financially supported by the Universiti Teknologi Malaysia GUP Grant 04H00 and the University of Malaya-Ministry of Higher Education High Impact Research Grant (UM MOHE HIR Grant No. H-50001-A000027). S.J. thanks the Slovak Grant Agency VEGA for financial support with grant No. 2/0150/14.
Author information
Authors and Affiliations
Contributions
S.J., S.S., R.N.Z.R.A.R., K.G.C. and K.M.G. conceived and designed the experiments. V.R., K.P.C. and K.G.C. performed the experiments. V.R., S.J., S.S. and K.M.G. analyzed the data. S.J., R.N.Z.R.A.R., K.G.C. and K.M.G. contributed reagents/materials/analysis tools. V.R., S.J., K.P.C. and K.M.G. wrote the paper. V.R., S.J., K.P.C. and K.M.G. prepared figures 1–5. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Ranjani, V., Janeček, Š., Chai, K. et al. Protein engineering of selected residues from conserved sequence regions of a novel Anoxybacillus α-amylase. Sci Rep 4, 5850 (2014). https://doi.org/10.1038/srep05850
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05850
This article is cited by
-
A novel low temperature active maltooligosaccharides-forming amylase from Bacillus koreensis HL12 as biocatalyst for maltooligosaccharide production
3 Biotech (2022)
-
Production and structure prediction of amylases from Chlorella vulgaris
Environmental Science and Pollution Research (2021)
-
Introduction of novel thermostable α-amylases from genus Anoxybacillus and proposing to group the Bacillaceae related α-amylases under five individual GH13 subfamilies
World Journal of Microbiology and Biotechnology (2018)
-
Crystal structure of a raw-starch-degrading bacterial α-amylase belonging to subfamily 37 of the glycoside hydrolase family GH13
Scientific Reports (2017)
-
Crystal structure of Anoxybacillus α-amylase provides insights into maltose binding of a new glycosyl hydrolase subclass
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
