
Abstract
In this study, nano-hydroxyapatite scaffolds with high mechanical strength and an interconnected porous structure were prepared using NTSS for the first time. The first step was performed using a laser characterized by the rapid heating to skip the surface diffusion and to obtain the driving force for grain boundary diffusion. Additionally, the interconnected porous structure was achieved by SLS. The second step consisted of isothermal heating in a furnace at a lower temperature (T2) than that of the laser beam to further increase the density and to suppress grain growth by exploiting the difference in kinetics between grain-boundary diffusion and grain-boundary migration. The results indicated that the mechanical properties first increased and then decreased as T2 was increased from 1050 to 1250°C. The optimal fracture toughness, compressive strength and stiffness were 1.69 MPam1/2, 18.68 MPa and 245.79 MPa, respectively. At the optimal point, the T2 was 1100°C, the grain size was 60 nm and the relative density was 97.6%. The decrease in mechanical properties was due to the growth of grains and the decomposition of HAP. The cytocompatibility test results indicated that cells adhered and spread well on the scaffolds. A bone-like apatite layer formed, indicating good bioactivity.
Similar content being viewed by others


Introduction
Bone tissue engineering offers an alternative approach for repairing bone defects caused by trauma, malignancies and congenital diseases1. Scaffolds, which are one of the key factors for tissue engineering, should possess sufficient mechanical strength to provide structural support and a porous structure to guide new bone tissue in-growth2,3. Moreover, the scaffolds should also possess good biocompatibility and osteoconductivity4,5. Hydroxyapatite (HAP) is widely used as a scaffold material because of its excellent osteoconductive properties and the bonding ability6,7,8. However, the inherent brittleness and low fracture toughness of the ceramics restrict their use in load-bearing applications9.
It is well known that the combination of an ultrafine grain size (such as nanoscale grain size) and a high density enable to improve the mechanical properties of ceramics to be improved10. However, preparing dense nano-grain ceramics using the conventional sintering method is difficult. The reason for this difficulty is that densification and grain growth are both driven by diffusive mechanisms, which results in the simultaneous activation of densification and grain growth11,12. Two-step sintering (TSS) is a promising approach for simultaneously obtaining high density and nano-grains13. Lukić et al.14 applied TSS to sinter HAP powder, which was conducted in a tube furnace. The relative density and average grain size reached 99.2% and 75 nm, respectively. Li et al.15 used TSS, which consisted of spark plasma sintering (SPS) as the first step and furnace sintering as the second step, to sinter TiO2 powder. The relative density and average grain size were 97.7% and 40 nm, respectively. However, these methods require sophisticated and expensive equipment and importantly, they lack the ability to precisely control the pore size, geometry, connectedness and spatial distribution16,17,18.
Selective laser sintering (SLS), as one type of the rapid prototyping (RP) technology, can obtain nano-size grains through the rapid heating provided by a high-energy laser beam19,20. This technology can also fabricate customized shapes and controlled internal architectures21,22. However, it is difficult to obtain the high densities due to the short action time (0.2–200 ms) between the laser beam and materials during the sintering process. In this study, we employ SLS as the first step for fabricating the interconnected porous structure in HAP scaffolds. Then, we use isothermal sintering at a lower temperature as the second step to keep the grain size at the nanoscale and to simultaneously improve the density through using grain-boundary diffusion. To be exact, this method first uses the high-energy laser beam to rapidly skip the surface diffusion and to achieve a sufficient driving force for grain-boundary diffusion, which is followed by isothermal sintering at a lower temperature to suppress grain-boundary migration and to keep the grain-boundary diffusion active.
This study was focused on preparing nano-HAP scaffolds with good mechanical properties and an interconnected porous structure via a novel two-step sintering (NTSS) process. The effects of T2 on the phase composition, relative density, grain size and mechanical properties were studied. The bioactivity was investigated by soaking the scaffolds in simulated body fluid (SBF). Moreover, human osteoblast-like MG-63 cells were cultured on the scaffolds and their adhesion and proliferation were examined.
Results
The nano-HAP powder possessed a needle-like shape and a uniform morphology (Figure 1a). A scaffold fabricated using NTSS is shown in Figures 1 (b–e). The scaffold was approximately 27 mm × 12 mm × 5 mm in size, with struts of approximately 1.6 mm in width, a pore size of approximately 800 μm and a wall thickness of approximately 800 μm.

SEM images and optical photographs.
(a) SEM image of the nano-HAP powder. Optical photographs (b) and SEM images (c–e) of the scaffold.
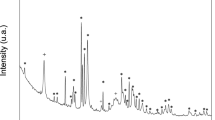
The XRD patterns of the nano-HAP powder and scaffolds prepared using SLS and NTSS are shown in Figure 2. All of the peaks matched with the JCPDS pattern 09-0432 for HAP, which suggested that no other phases were present, as shown in Figure 2 (a). The XRD pattern of the scaffolds prepared using SLS without the second sintering step is shown in Figure 2 (b). All of the peaks were attributed to the HAP phase and no additional peaks were observed. The results indicated that the HAP did not decompose after laser sintering. The XRD patterns of the scaffolds prepared via the NTSS at T2 of 1050, 1100, 1150, 1200 and 1250°C are shown in Figures 2 (c), (d), (e), (f) and (g), respectively. The peaks became narrower and sharper as the sintering temperature increased. A peak shift could be observed when comparing the pattern with that of the initial nano-HAP powder, which indicated that the HAP lattice had contracted due to the dehydroxylation of HAP23. There were no phase transitions in the scaffolds sintered at temperatures of 1050°C and 1100°C. However, a small amount of HAP was decomposed into β-TCP and TTCP when the temperatures were 1150°C and 1200°C, moreover, small amounts of α-TCP occurred at the temperature of 1250°C.

XRD patterns.
(a) The nano-HAP powder. (b) The SLS scaffold without the second sintering step. (c–g) The scaffolds sintered at different T2.
The FTIR spectra of the nano-HAP powder, of the scaffolds prepared using SLS without the second sintering step and of scaffolds prepared using NTSS at different T2 are shown in Figure 3. In the FTIR spectra, the bands at 3570 and 630 cm−1 were attributed to the hydroxyl stretching bands and bending bands of HAP, respectively24. The broad absorption band from 3600 to 3300 cm−1 indicated the existence of the bending mode of absorbed water25. The bands at 1093 and 1041 cm−1 were assigned to the components of the triply degenerate ν3 antisymmetric P-O stretching mode and the ν1 P-O symmetric stretching mode was detected at 962 cm−126,27. The bands at 603 and 569 cm−1 were attributed to components of the triply degenerate ν4 O-P-O bending mode and the doubly degenerate ν2 O-P-O bending mode was evident at 470 cm−128,29. The relative intensities of the OH− vibration bands decreased as the sintering temperature increased, indicating the occurrence and development of dehydroxylation. Bands corresponding to OH groups and to PO43− were still present when the sintering temperature reached to 1250°C, which indicated that the HAP experienced very limited phase decomposition during the NTSS process.

FTIR spectra.
(a) The nano-HAP powder. (b) The SLS scaffold without the second step sintering. (c–g) The scaffolds sintered at different T2.
To obtain ultrafine grains and high densities, the scaffolds were fabricated using NTSS. The first step was conducted using SLS under the preset parameters. For this step, the temperature of the laser beam was greater than 1250°C30 and the relative density was 82.6%. Subsequently, the relative densities increased during the second step (Figure 4). The relative density rapidly increased from 91.2% at 1050°C to 97.6% at 1100°C and then it slowly increased to 97.9% at 1150°C. However, the relative density slightly decreased when T2 was 1200°C or greater, which results from the decomposition of HAP to TCP. As is well known, β-TCP and α-TCP have densities of 3.07 and 2.86 g/cm3, respectively, which are lower than that of HAP (the theoretical density of HAP is 3.16 g/cm3)31,32, respectively.

Relative densities of the struts sintered using NTSS at different T2.
The microstructures of the scaffolds without and with the second sintering step are shown in Figures 5 (a–e). It could be observed that the scaffold had an incompact structure and that some pores were unevenly distributed (Figure 5a). Slow grain growth occurred when the scaffold was sintered at 1100°C (Figure 5b). The grain growth was evident when the temperature was 1150°C or greater (Figures 5c–e). The average grain sizes of the scaffolds prepared using SLS without the second sintering step and of those prepared using NTSS at different T2 are shown in Figure 5 (f). The average grain size was approximately 50 nm when the scaffolds prepared using SLS without the second sintering step. The average grain size increased from 56 nm to 160 nm when T2 was increased from 1050°C to 1250°C for the scaffolds prepared using NTSS.

The etched surfaces of the scaffolds without the second sintering step (a); sintered at T2 of 1100 (b) °C, 1150°C (c), 1200°C (d) and 1250°C (e); and the average grain sizes of the scaffolds prepared using SLS without the second sintering step and of those using NTSS at different T2 (f).
The mechanical properties of the scaffolds without the second sintering step and of those sintered at different T2 are shown in Figure 6. The Vickers hardness, fracture toughness, compressive strength and stiffness of the scaffold without the second sintering step were 3.45 GPa, 1.11 MPam1/2, 6.45 MPa and 168.24 MPa, respectively. The mechanical properties of the scaffolds prepared using NTSS increased as the temperature was increased to 1100°C and subsequently decreased as the temperature was increased from 1150 to 1250°C. The optimal Vickers hardness, fracture toughness, compressive strength and stiffness were 4.63 GPa, 1.69 MPam1/2, 18.68 MPa and 245.79 MPa, respectively. The mechanical properties at 1100°C were markedly higher than those without the second sintering step (P < 0.001).

Vickers hardness and fracture toughness (a) and compressive strength and stiffness (b) of the SLS scaffold without the second sintering step and of scaffolds sintered at different T2 (1050, 1100, 1150, 1200 and 1250°C).
Difference (*P < 0.05 and **P < 0.001) between the scaffolds prepared using NTSS compared to those without the second sintering step.
The surface morphologies of the scaffolds after soaking in SBF for various lengths of time are shown in Figure 7. Numerous tiny agglomerates, which were attributed to apatite nuclei, could be observed on the surface after 2 days of soaking (Figures 7a, b). The quantities and sizes of these precipitates increased as the soaking time increased to 7 days (Figures 7c, d). The surface was completely covered by a layer of flake-like precipitates after 14 days of soaking (Figures 7e, f). The layer became denser and was composed of many elongated and interlaced, worm-like, superfine crystallites as the soaking time increased to 21 days (Figures 7g, h).

SEM micrographs of the scaffolds soaked in SBF for 2 (a, b), 7 (c, d), 14 (e, f) and 21 (g, h) days; (b), (d), (f) and (h) are higher magnifications images of those presented in (a), (c), (e) and (g), respectively.
The SEM-EDS spectra of the scaffolds after soaking in SBF for 0, 2, 7, 14 and 21 days are shown in Figures 8 (a–e). It could be observed that the precipitates were calcium- and phosphorus-rich phases, which also contained carbon and oxygen. Small amounts of Na, Cl and Mg, which were derived from the SBF solution, were detected. The changing trend in Ca/P ratios is shown in Figure 8 (f). The Ca/P ratio of the scaffolds before soaking in SBF was 1.67. The Ca/P ratios slightly increased to 1.79 as the soaking time increased.

EDS analysis for the scaffolds soaked in SBF for 0 (a), 2 (b), 7 (c), 14 (d) and 21 (e) days; changing trend of Ca/P ratios (f) on the surface of scaffolds after soaking in SBF.
The morphologies of MG-63 cells cultured on the scaffolds for 4 hours and for 1, 3 and 5 days are shown in Figure 9. The numbers of cells attached to the scaffolds increased with the culture time. The MG-63 cells anchored tightly on the surface of the scaffolds and exhibited a fusiform-shape after 4 hours of culture. Significant spreading of viable cells with extended fibers was observed along the surface of the scaffolds after 1 day. After 3 days, the cells grew and spread out on the surface and neighbor cells connected with each other through cytoplasmic extensions. The cells were attached across the entire surfaces of the scaffolds and some of them formed bridges and formed a dense cellular construct that was clearly visible after 5 days.

SEM images of MG-63 cells cultured on the scaffolds for 4 hours (a) and for 1 (b), 3 (c) and 5 (d) days.
Discussion
The scaffold possessed a well-ordered three-dimensional (3D) architecture and interconnected pore channels (Figure 1). The open and interconnected pore network was a crucial factor for the scaffold to allow cell growth and the transport of nutrients and metabolic waste. Gotz et al.33 reported that pore sizes of >300 μm were recommended for implants due to enhanced new bone and capillary formation. Hollister et al.34 conducted in vivo studies on HAP scaffolds with pore diameters ranging between 400 μm and 1,200 μm and observed that bone growth on scaffolds for all pore sizes, with no significant difference between pore sizes.
In the present study, the thermal change of pure HAP is reported to occur in two stages: dehydroxylation and decomposition35,36. During dehydroxylation, HAP molecules lost OH radicals during heating at high temperatures and oxyapatite formed according to the following equation37,38:


where Ca10(PO4)6(OH)2 is HAP, Ca10(PO4)6O is oxyapatite and X is a non-charged vacancy; one of the lattice sites that were originally occupied by two OH groups in a HAP unit cell was now occupied by an oxygen atom while leaving the other vacant, which generally caused lattice contraction. HAP could decompose into TCP and TTCP with increasing sintering temperature through the following process39,40:

where Ca3(PO4)2 is TCP and Ca4P2O9 is TTCP. The presence of α-TCP was due to the transformation of β-TCP to α-TCP.
The crystallite size of the scaffold prepared using NTSS at 1100°C in the direction perpendicular to a certain crystal face was estimated using the Scherrer equation41:

where D is the crystallite size, λ is the wavelength of Cu Kα radiation (λ = 1.54056 Å), β is the full width of the peak at half of the maximum intensity (FWHM) and θ is the diffraction angle of the corresponding reflection.
The crystallite sizes estimated from the (002) and (300) reflection peak were 56.54 and 64.32 nm, respectively, as calculated from Scherrer equation. The average grain size of the scaffold prepared through NTSS at 1100°C was approximately 60 nm from the SEM image (Figure 5). The calculated result from Scherrer equation conformed to the measured result from SEM.
As shown in Figures 4, 5 and 6, the Vickers hardness, fracture toughness, compressive strength and stiffness first increased from 4.12 GPa, 1.35 MPam1/2, 14.28 MPa and 224.51 MPa to 4.63 GPa, 1.69 MPam1/2, 18.68 MPa and 245.79 MPa, respectively, as the relative density increased from 91.2% to 97.9%. Then, these values decreased from 4.63 GPa, 1.69 MPam1/2, 18.68 MPa and 245.79 MPa to 3.63 GPa, 1.41 MPam1/2, 11.76 MPa and 194.91 MPa, respectively, as the grain size increased from 60 nm to 160 nm. It could be observed that the mechanical properties of the HAP ceramic were closely related with its density and grain size. The mechanical properties increased primarily because of the increase in density when T2 was increased from 1050 to 1100°C, whereas they decreased primarily because of the combined effects of the increase in grain size and the decomposition of HAP when T2 was increased from 1150 to 1250°C. Several studies have reported that a finer grain size would result in greater strength and toughness in comparison with coarser grain sizes42,43,44. Additionally, some studies45,46,47 have reported that the decomposition of HAP would decrease its mechanical properties. The results of the phase composition, relative density, microstructure and mechanical property analyses implied that NTSS is an effective method for fabricating scaffolds with good mechanical properties. The optimum T2 for the NTSS method was 1100°C.
The presence of carbon in the apatite implied that a bone-like apatite layer was formed on the surfaces of scaffolds after soaking in SBF (Figure 8). Previous studies have shown that the formation of a bone-like apatite layer on the surface of biomaterials in SBF plays an important role in improving osteoblast growth and differentiation, which further influences their in vivo bone-forming ability48,49,50. The increase in Ca/P ratios was due to the substitution of phosphate groups by carbonate groups during apatite formation51. The scaffolds were effective in promoting cellular adhesion, spreading, proliferation and formation of extracellular matrix protein (ECM) such that the cells merged on the surfaces of scaffolds forming a layer or sheet (Figure 9). These results indicated that the scaffolds provided good biocompatibility for cell attachment, proliferation and viability.
In summary, dense nano-HAP scaffolds with an interconnected porous structure for bone tissue engineering were successfully fabricated using NTSS. The NTSS method used SLS as the first step, followed by isothermal sintering as the second step. The average grain size increased from 56 to 160 nm as T2 increased from 1050 to1250°C. The relative density increased from 91.2% at 1050°C to 97.9% at 1150°C and then decreased to 96.8% at 1250°C. HAP decomposed into TCP and TTCP when the sintering temperature was 1150°C or greater. The Vickers hardness, fracture toughness, compressive strength and stiffness first increased and subsequently decreased. The scaffold with optimal mechanical properties was fabricated at a T2 of 1100°C; at this point, the average grain size was 60 nm and the relative density was 97.6%. The formation of a bone-like apatite layer in SBF suggested good bioactivity. Moreover, the attachment and proliferation of MG-63 cells on the scaffolds indicated good cytocompatibility.
Methods
Materials and experiments
The nano-HAP powder (No. MHAP04) was obtained from the Nanjing Emperor Nano Material Co., Ltd and it was synthesized using the sol-gel technique with Ca(NO3)2·4H2O and (CH3O)3P as precursors. The powder had a width of 20 nm, a length of 150 nm, a purity of 99.5%, a whiteness of 96 and a pH of 7.41.
The sintering profile is shown in Figure 10. The scaffolds were first rapidly sintered to a high temperature (T1) by SLS and then rapidly cooled to room temperature. Subsequently, the scaffolds were heated at 1°C/min to a temperature (T2) for isothermal sintering and after a dwell time of 3 h, they were cooled to 400°C at a cooling rate of 1°C/min; finally, the scaffolds were slowly cooled to room temperature within the furnace.

Sintering profiles for NTSS and TSS.
The first step sintering was performed using on a homemade SLS system equipped with a 100 W CO2 laser (model Firestar® t-Series, Synrad Co., USA) with a wavelength of 10.6 μm and a minimum laser spot diameter of 50 μm52. Briefly, a layer of the nano-HAP powder was deposited on the elevating platform. The deposited layer was selectively radiated by the laser beam and bonded to form a solidified layer. The elevating platform was moved down by an amount equivalent to the thickness of a layer. By repeating the above steps, a number of solidified layers were stacked to construct the scaffold. The un-bonded powder was removed with water. The second sintering step was performed under different T2 in a furnace (JNL-16XB, Luoyang Liyu Co., LTD. China). The NTSS processing parameters are shown in Table 1.
Scaffold characterization
The morphologies of the starting powder and of the scaffolds were observed by scanning electron microscopy (SEM) (Tescan, Mira 3 FEG-SEM, TESCAN Co., Czech) at an accelerating voltage of 5.0 kV. The samples were sputter coated with a thin layer of gold/palladium for 120 s using an auto fine coater (JFC-1600, JEOL, Ltd., Japan) before SEM examination. The struts samples (3 × 1.6 × 0.8 mm3) were polished using diamond paste to a 1 mm surface finish and then ultrasonically cleaned. The microstructure was revealed by chemical etching with 5% hydrofluoric acid (HF) for 2 min and examined using SEM (JEOL JSM-5600LV, JEOL Ltd., Japan). The average grain size was measured from SEM images using the linear intercept method.
The phase composition was analyzed by X-ray diffraction (XRD) (D8-ADVANCE, German) with Cu Kα radiation (40 kV, 40 mA). The analysis was made with a scanning speed of 8°/min, a step size of 0.02° over the 2θ range of 20–50° at room temperature. Prior to analysis, the scaffolds were ground to a fine powder using a pestle and mortar. The phases were identified by comparing the X-ray diffractograms with standards patterns complied by the Joint Committee on Powder Diffraction Standards (JCPDS) using cards 09-0432 for HAP, 09-0169 for β-TCP, 09-0348 for α-TCP and 25-1137 for tetracalcium phosphate (TTCP). The functional groups were determined by Fourier transform infrared spectroscopy (FTIR) (NicoletteTM 6700, Thermo Electron Corp., USA) over a wavenumber range of 400–4000 cm−1 and with a resolution of 2 cm−1. The scaffolds were ground into a powder in an agate mortar and thoroughly mixed with KBr. Transparent pellets were prepared in a stainless steel die by applying a uniaxial load of 1000 psi.
The mass of the scaffold samples (15 × 15 × 7 mm3) were measured using an electronic balance. The volumes of the scaffolds were measured using the Archimedes' method with distilled water. The apparent density was calculated as the mass divided by the volume, which was the actual density of the strut. The relative density was calculated as the actual density divided by theoretical density of HAP (3.16 g/cm3). Six measurements were conducted to obtain the average value. The Vickers hardness and fracture toughness (KIC) of the strut samples (3 × 1.6 × 0.8 mm3) were obtained by performing indentation tests with a Vickers microindenter (HXD-1000TM/LCD, Digital Micro Hardness Tester, Shanghai Taiming Optical Instrument Co. Ltd, China). Prior to indentation, all of the strut samples were vertically inlaid in epoxy with an inlaying machine (XQ-2B, Φ22*15). Then, the surface of the sample was polished using a grinding & polishing machine (Model MP-2, Laizhou Weiyi Experiment Machine Manufacturing Co., Ltd, China) and subjected to indentation. The indentation load was maintained constant at 2.94 N and a loading time of 15 s was employed. The KIC was calculated using the following formula which was derived from the model proposed by Evans and Charles53: KIC = 0.0824Pc−3/2, where P is the applied indentation load and c is the length of the induced radial crack. The compressive strength and stiffness were determined by axially crushing the scaffolds (15 × 15 × 7 mm3) between two flat plates using a microcomputer control electronic universal test machine (WD-D1, Shanghai zhuoji Instruments Co. Ltd, China) with a loading rate of 0.5 mm/min. The stress versus strain responses of the scaffolds were simultaneously collected by data collection software on a computer and saved in a spreadsheet. By curve fitting with a linear function of the stress-strain relation during the loading stage in the region of the compression peak, the slope of the stress-strain curve, which represents the stiffness, could be determined. Six samples of each type were tested to obtain the average values, along with their standard deviations, for the mechanical properties, relative density and average grain size.
Assessment of biological properties
SBF, which had ion concentrations (142.0 mM Na+, 5.0 mM K+, 1.5 mM Mg2+, 2.5 mM Ca2+, 148.3 mM Cl−, 4.2 mM HCO3−, 1.0 mM HPO42− and 0.5 mM SO42−) and a pH value (7.4) similar to those of human blood plasma, was prepared in accordance with the procedure described by Kokubo and co-workers54. The scaffolds samples (15 × 15 × 7 mm3) were soaked in SBF at 37°C for 2, 7, 14 and 21 days and the solutions were refreshed daily. The scaffolds were removed from the SBF after incubation for the specified time periods, gently rinsed with distilled water and then dried in an electrothermal blowing dry box (101-00S, Guangzhou Dayang Electronic Machinery Equipment Co., Ltd, China) at 37°C for 1 day before further characterization. A SEM equipped with an energy-dispersive X-ray spectrometer (EDS) was used to evaluate the surface morphologies of the scaffolds after immersion in SBF, as well as to identify the formation of apatite.
The MG-63 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 5% penicillin/streptomycin and 5% glutamine and they were incubated at 37°C under 5% CO2 and maintained according to the cell line-specific recommendations of the ATCC. All of the scaffolds samples (15 × 15 × 7 mm3) were immersed in the cell culture medium for 24 h in 24-well tissue culture plates, which had been sterilized in 75% ethanol for 10 min and immersed in phosphate-buffered saline (PBS) overnight. Then, the MG-63 cells were seeded onto the prewetted scaffolds (2 × 105 cells/scaffold). The cell/scaffold constructs were placed in 24-well tissue culture plates and incubated at 37°C under 5% CO2 for various periods of time (4 hours and 1, 3 and 5 days) and the medium was exchanged daily. At each culture period, the scaffolds were removed and transferred to new 24-well tissue culture plates. After being washed twice with PBS solution, they were dehydrated in a graded ethanol series for 10 min and critically point dried. Additionally, the scaffolds were gold sputtered under vacuum and observed by SEM.
Statistical analysis
Data for the Vickers hardness, fracture toughness, compressive strength and stiffness are presented as the mean ± standard deviation. Statistical analysis was performed using SPSS (Statistical Package for the Social Sciences) software (SPSS Statistics version 19, IBM, Armonk, NY, USA). A value of *P < 0.05 was considered to be statistically significant and **P < 0.001 was considered to be remarkably significant.
References
Gu, Y., Huang, W., Rahaman, M. N. & Day, D. E. Bone regeneration in rat calvarial defects implanted with fibrous scaffolds composed of a mixture of silicate and borate bioactive glasses. Acta. Biomater. 9, 9126–9136 (2013).
Zhang, X. et al. Polymer-ceramic spiral structured scaffolds for bone tissue engineering: effect of hydroxyapatite composition on human fetal osteoblasts. Plos. One. 9, e85871 (2014).
Meng, J. et al. Super-paramagnetic responsive nanofibrous scaffolds under static magnetic field enhance osteogenesis for bone repair in vivo. Sci. Rep. 3, 2655 (2013).
Ibara, A. et al. Osteoconductivity and biodegradability of collagen scaffold coated with nano-β-TCP and fibroblast growth factor 2. J. Nanomater. 2013, 46 (2013).
Jin, L., Wan, Y., Shimer, A. L., Shen, F. H. & Li, X. J. Intervertebral disk-like biphasic scaffold-demineralized bone matrix cylinder and poly (polycaprolactone triol malate)-for interbody spine fusion. J. tissue. eng. 3, 2041731412454420 (2012).
Lin, K., Chen, L. & Chang, J. Fabrication of dense hydroxyapatite nanobioceramics with enhanced mechanical properties via two-step sintering process. Int. J. Appl. Ceram. Tec. 9, 479–485 (2012).
Malina, D., Biernat, K. & Sobczak-Kupiec, A. Studies on sintering process of synthetic hydroxyapatite. Acta. Biochim. Pol. 60, 851–855 (2012).
Kim, B. S., Kang, H. J., Yang, S. S. & Lee, J. Comparison of in vitro and in vivo bioactivity: cuttlefish-bone-derived hydroxyapatite and synthetic hydroxyapatite granules as a bone graft substitute. Biomed. Mater. 9, 025004 (2014).
Eshraghi, S. & Das, S. Micromechanical finite-element modeling and experimental characterization of the compressive mechanical properties of polycaprolactone-hydroxyapatite composite scaffolds prepared by selective laser sintering for bone tissue engineering. Acta. Biomater. 8, 3138–3143 (2012).
Gallardo-López, A., Domínguez-Rodríguez, A., Estournès, C., Marder, R. & Chaim, R. Plastic deformation of dense nanocrystalline yttrium oxide at elevated temperatures. J. Eur. Ceram. Soc. 32, 3115–3121 (2012).
Mazaheri, M., Zahedi, A. M. & Sadrnezhaad, S. K. Two-step sintering of nanocrystalline ZnO compacts: effect of temperature on densification and grain growth. J. Am. Ceram. Soc. 91, 56–63 (2008).
Mazaheri, M., Zahedi, A. M., Haghighatzadeh, M. & Sadrnezhaad, S. K. Sintering of titania nanoceramic: densification and grain growth. Ceram. Int. 35, 685–691 (2009).
Isobe, T., Ooyama, A., Shimizu, M. & Nakajima, A. Pore size control of Al2O3 ceramics using two-step sintering. Ceram. Int. 38, 787–793 (2012).
Lukić, M. J., Škapin, S. D., Marković, S. & Uskoković, D. Processing route to fully dense nanostructured HAp Bioceramics: From powder synthesis to sintering. J. Am. Ceram. Soc. 95, 3394–3402 (2012).
Li, B., Liu, D., Liu, J., Hou, S. & Yang, Z. Two-step sintering assisted consolidation of bulk titania nano-ceramics by spark plasma sintering. Ceram. Int. 38, 3693–3699 (2012).
Mazaheri, M., Haghighatzadeh, M., Zahedi, A. M. & Sadrnezhaad, S. K. Effect of a novel sintering process on mechanical properties of hydroxyapatite ceramics. J. alloy. compd. 471, 180–184 (2009).
Mazaheri, M., Simchi, A. & Golestani-Fard, F. Densification and grain growth of nanocrystalline 3Y-TZP during two-step sintering. J. Eur. Ceram. Soc. 28, 2933–2939 (2008).
Alvarez, K. & Nakajima, H. Metallic scaffolds for bone regeneration. Materials 2, 790–832 (2009).
Shuai, C. et al. Structure and properties of nano-hydroxypatite scaffolds for bone tissue engineering with a selective laser sintering system. Nanotechnology 22, 285703 (2011).
Shishkovsky, I., Morozov, Y. & Smurov, I. Nanofractal surface structure under laser sintering of titanium and nitinol for bone tissue engineering. Appl. Surf. Sci. 254, 1145–1149 (2007).
Zhang, Y. et al. In vitro biocompatibility of hydroxyapatite-reinforced polymeric composites manufactured by selective laser sintering. J. Biomed. Mater. Res. A 91, 1018–1027 (2009).
Duan, B. et al. Three-dimensional nanocomposite scaffolds fabricated via selective laser sintering for bone tissue engineering. Acta. Biomater. 6, 4495–4505 (2010).
Lin, C., Xiao, C. & Shen, Z. Nano pores evolution in hydroxyapatite microsphere during spark plasma sintering. Sci. Sinter. 43, 39–46 (2011).
Balgová, Z., Palou, M., Wasserbauer, J., Lutišanová, G. & Kozánkováb, J. Preparation, characterization and in vitro bioactivity of polyvinyl alcohol-hydroxyapatite biphasique membranes. Acta. Chimica. Slovaca. 6, 8–14 (2013).
Chen, L., Mccrate, J. M., Lee, J. C. & Li, H. The role of surface charge on the uptake and biocompatibility of hydroxyapatite nanoparticles with osteoblast cells. Nanotechnology 22, 105708 (2011).
Bose, S., Dasgupta, S., Tarafder, S. & Bandyopadhyay, A. Microwave-processed nanocrystalline hydroxyapatite: Simultaneous enhancement of mechanical and biological properties. Acta. Biomater. 6, 3782–3790 (2010).
Ribeiro, C. C., Gibson, I. & Barbosa, M. A. The uptake of titanium ions by hydroxyapatite particles-structural changes and possible mechanisms. Biomaterials 27, 1749–1761 (2006).
Viswanath, B. & Ravishankar, N. Controlled synthesis of plate-shaped hydroxyapatite and implications for the morphology of the apatite phase in bone. Biomaterials 29, 4855–4863 (2008).
Rapacz-Kmita, A., Paluszkiewicz, C., Ślósarczyk, A. & Paszkiewicz, Z. FTIR and XRD investigations on the thermal stability of hydroxyapatite during hot pressing and pressureless sintering processes. J. Mol. Struct. 744, 653–656 (2005).
Shuai, C., Feng, P., Gao, C., Zhou, Y. & Peng, S. Simulation of dynamic temperature field during selective laser sintering of ceramic powder. Math. Comp. Model. Dyn. 19, 1–11 (2013).
saito, E. et al. Periodontal repair following implantation of beta-tricalcium phosphate with different pore structures in class III furcation defects in dogs. Dent. Mater. J. 31, 681–688 (2012).
Ahmad, N. et al. Effect of precursor's solubility on the mechanical property of hydroxyapatite formed by dissolution-precipitation reaction of tricalcium phosphate. Dent. Mater. J. 31, 995–1000 (2012).
Gotz, H. E. et al. Effect of surface finish on the osseointegration of laser-treated titanium alloy implants. Biomaterials 25, 4057–4064 (2004).
Hollister, S. J. et al. Engineering craniofacial scaffolds. Orthod. Craniofac. Res. 5, 162–173 (2005).
Zhang, Y., Lu, J. & Yang, S. Preparation of hydroxyapatite ceramic through centrifugal casting process using ultra-fine spherical particles as precursor and its decomposition at high temperatures. J. Adv. Ceram. 1, 60–65 (2012).
Hannora, A. E., Mukasyan, A. S. & Mansurov, Z. A. Nanocrystalline hydroxyapatite/Si coating by mechanical alloying technique. Bioinorg. Chem. Appl. 2012, 390104 (2012).
Ye, H., Liu, X. Y. & Hong, H. Characterization of sintered titanium/hydroxyapatite biocomposite using FTIR spectroscopy. J. Mater. Sci-Mater. M. 20, 843–850 (2009).
Muralithran, G. & Ramesh, S. The effects of sintering temperature on the properties of hydroxyapatite. Ceram. Int. 26, 221–230 (2000).
Liu, H. S. et al. Hydroxyapatite synthesized by a simplified hydrothermal method. Ceram. Int. 23, 19–25 (1997).
Marcelo, T. M., Livramento, V., de Oliveira, M. V. & Carvalho, M. H. Microstructural characterization and interactions in Ti- and TiH2-hydroxyapatite vacuum sintered composites. Mater. Res. 9, 65–71 (2006).
Rajkumar, M., Sundaram, N. M. & Rajendran, V. Preparation of size controlled, stoichiometric and bioresorbable hydroxyapatite nanorod by varying initial pH, Ca/P ratio and sintering temperature. Dig. J. Nanomater. Bios. 6, 169–179 (2011).
Mazaheri, M., Valefi, M., Hesabi, Z. R. & Sadrnezhaad, S. K. Two Step sintering of titania nanoceramics assisted by anatase to rutile phase transformation. Ceram. Int. 35, 13–20 (2009).
Ramesh, S., Tan, C. Y., Sopyan, I., Hamdi, M. & Teng, W. D. Consolidation of nanocrystalline hydroxyapatite powder. Sci. Technol. Adv. Mater. 8, 124–130 (2007).
Armstrong, R. W. Grain size dependent alumina fracture mechanics stress intensity. Int. J. Refract. Met. H. 19, 251–255 (2001).
Li, S., Izui, H., Okano, M. & Watanabe, T. The effects of sintering temperature and pressure on the sintering behavior of hydroxyapatite powder prepared by spark plasma sintering. J. Biomech. Eng. 3, 1–12 (2008).
Khalil, K. A., Won Kim, S. & Kim, H. Y. Consolidation and mechanical properties of nanostructured hydroxyapatite-(ZrO2 + 3 mol% Y2O3) bioceramics by high-frequency induction heat sintering. Mat. Sci. Eng. A-Struct. 456, 368–372 (2007).
Gu, Y. W., Loh, N. H., Khor, K. A., Tor, S. B. & Cheang, P. Spark plasma sintering of hydroxyapatite powders. Biomaterials 23, 37–43 (2002).
Yu, L. et al. A novel injectable calcium phosphate cement-bioactive glass composite for bone regeneration. PloS. One. 8, e62570 (2013).
Xu, S. et al. Reconstruction of calvarial defect of rabbits using porous calcium silicate bioactive ceramics. Biomaterials 29, 2588–2596 (2008).
Wu, C., Luo, Y., Cuniberti, G., Xiao, Y. & Gelinsky, M. Three-dimensional printing of hierarchical and tough mesoporous bioactive glass scaffolds with a controllable pore architecture, excellent mechanical strength and mineralization ability. Acta. Biomater. 7, 2644–2650 (2011).
Rohanizadeh, R., Trecant-Viana, M. & Daculsi, G. Ultrastructural study of apatite precipitation in implanted calcium phosphate ceramic: influence of the implantation site. Calcified. Tissue. Int. 64, 430–436 (1999).
Shuai, C. et al. Correlation between properties and microstructure of laser sintered porous β-tricalcium phosphate bone scaffolds. Sci. Technol. Adv. Mater. 14, 055002 (2013).
Evans, A. G. & Charles, E. A. Fracture toughness determination by indentation. J. Am. Ceram. Soc. 59, 371–372 (1976).
Kokubo, T. et al. Ca, P-rich layer formed on high-strength bioactive glass-ceramic A-W. J. Biomed. l Mater. Res. 24, 331–343 (1990).
Acknowledgements
This work was supported by the following funds: (1) The Natural Science Foundation of China (51222506, 81372366); (2) Hunan Provincial Natural Science Foundation of China (14JJ1006); (3) Program for New Century Excellent Talents in University (NCET-12-0544); (4) The Fundamental Research Funds for the Central Universities (2011JQ005, 2012QNZT015); (5) Project supported by the Fok Ying-Tong Education Foundation, China (131050); (6) Shenzhen Strategic Emerging Industrial Development Funds (JCYJ20130401160614372); (7) The Open-End Fund for the Valuable and Precision Instruments of Central South University; (8) The Fundamental Research Funds for the Central Universities of Central South University; (9) The faculty research grant of Central South University (2013JSJJ011, 2013JSJJ046).
Author information
Authors and Affiliations
Contributions
P.F. and C.S. conceived and designed the research. P.F., M.N., C.G. and S.P. performed experiments. P.F., M.N. and C.S. analyzed the data and wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Feng, P., Niu, M., Gao, C. et al. A novel two-step sintering for nano-hydroxyapatite scaffolds for bone tissue engineering. Sci Rep 4, 5599 (2014). https://doi.org/10.1038/srep05599
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05599
This article is cited by
-
Advancement in “Garbage In Biomaterials Out (GIBO)” concept to develop biomaterials from agricultural waste for tissue engineering and biomedical applications
Journal of Environmental Health Science and Engineering (2022)
-
Fabrication of Nanohydroxyapatite-Chitosan Coatings by Pulse Electrodeposition Method
Journal of Inorganic and Organometallic Polymers and Materials (2022)
-
Laser sintering of coated polyamide 12: a new way to improve flammability
Journal of Materials Science (2022)
-
Microstructural, electrical and biological activity in \(\mathrm{Ca}_{10}(\mathrm{PO}_4)_6(\mathrm{OH})_2-\mathrm{Ba}_{0.5}\mathrm{Sr}_{0.5}\mathrm{TiO}_3\) ceramic composites designed for tissue engineering applications
Scientific Reports (2021)
-
Selective laser melting of Zn-Si-substituted hydroxyapatite
Russian Chemical Bulletin (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
