
Abstract
Direct in vivo administration of messenger RNA (mRNA) delivered in both naked and nanoparticle formats are actively investigated because the use of dendritic cells transfected ex vivo with mRNA for cancer therapy is expensive and needs significant infrastructure. Notably, intravenous and subcutaneous injections are the only routes of administration tested for mRNA nanoparticle tumor vaccination. In this report, we demonstrate that tumor immunity can be achieved via nasal administration of mRNA. Mice nasally immunized with mRNA delivered in nanoparticle format demonstrate delayed tumor progression in both prophylactic and therapeutic immunization models. The observed tumor immunity correlates with splenic antigen-specific CD8+ T cells and is achieved only when mRNA is delivered in nanoparticle but not in naked format. In conclusion, we demonstrate, as a proof-of-concept, a non-invasive approach to mRNA tumor vaccination, increasing its potential as a broadly applicable and off-the-shelf therapy for cancer treatment.
Similar content being viewed by others
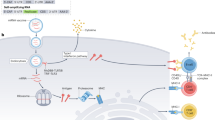


Introduction
Tumor vaccination employing mRNA transfected dendritic cells (DCs) has been shown to be an effective strategy for treatment of cancer1,2,3,4,5,6. Promising results emerging from recent clinical trials7,8,9 supports the notion that this is a strategy that can be translated to humans and is amenable to commercialization. However, this process involves harvesting cells from patients via leukapheresis, generating DCs in vitro from adherent monocytes, loading them with mRNA, maturing them in vitro and re-injecting these mRNA-loaded DCs back into the patient. This is a cost, labor and resource intensive procedure. Because of these reasons, researchers have explored alternative cell-based approaches10,11, as well as direct in vivo injection of mRNA in naked12,13 and nanoparticle formats14,15,16,17,18,19. However, due to rapid degradation of naked mRNA in vivo, direct injection of mRNA is effective only when it is injected directly into lymph nodes12,13. Intranodal injection is an extremely invasive procedure in mice and hence not amenable for repeated administrations. Moreover, although intranodal injection is performed using ultrasound-guidance in humans, it remains a technically challenging procedure that requires surgical expertise. While this approach is an improvement over the existing ex vivo DC-based approach, scale-up remains a significant challenge thus hindering its broad application.
A strategy that overcomes this problem is encapsulating mRNA in nanoparticles, which not only protects mRNA from nuclease degradation, but also facilitates its uptake by cells and endosome escape within cells leading to enhanced delivery efficiencies. This approach may obviate the need for intranodal administration, while still permitting direct in vivo application of an off-the-shelf mRNA vaccine formulation via conventional routes of administration. Indeed, mRNA nanoparticle delivery has attracted interest from many research groups in recent years17,20,21,22,23. In addition, therapeutic efficacy of mRNA encapsulated in nanoparticles for tumor vaccination has also been recently demonstrated16,17. Notably, tail vein and subcutaneous injections are the only routes evaluated in all of these studies. We have previously reported that primary DCs can be efficiently transfected by mRNA encapsulated in nanoparticles in vitro. These particles are about 180 nm and 300 nm in hydrodynamic diameter and have zeta potentials of +40 mV/−12 mV in water and 10% FBS supplemented media, respectively24. In the same study, we determined that luciferase expression mediated by nasally administered mRNA nanoparticles lasts for about 24 hours compared to naked mRNA, which is detectable only up to 4 hours post-administration24. The bioavailability of transgene product is clearly superior to nasally instilled soluble protein antigens, where >85% of the soluble antigen is cleared from the nasal site within 6 hours25.
In this study, we hypothesize that nasal vaccination could be an effective strategy for mRNA tumor vaccination. Intranasal route of immunization is desirable because of its non-invasive nature, amenability for repeated administration and is associated with high patient compliance. It has been previously reported that intranasal immunization with naked mRNA can induce a moderate level of protection against tuberculosis in mice26. We reason that significantly higher nasal transfection efficiencies mediated by mRNA nanoparticles could translate to the induction of anti-tumor immunity. In addition, a previous study has reported that intranasal tumor vaccination with soluble OVA peptides can induce robust anti-tumor immunity27. Therefore we reason that the prolonged presence of antigen at the nasal site where the Nasal-Associated Lymphoid Tissues (NALT) are located28 could translate to enhanced immune responses. Last but not least, we observe that nasally administered nanoparticles are taken up by CD11chigh cells isolated from NALT (Supplementary Figure S1), indicating that this route of administration could be used to directly target DCs.
Based on above rationale, we set up an immunization scheme to investigate the therapeutic efficacy (Figure 1) of chicken ovalbumin (OVA) encoding mRNA nanoparticle vaccination in prophylactic and therapeutic immunotherapy models with E.G7-OVA tumor cells. The immunization schemes are based on published prime-boost protocol that entails weekly nasal administration for three weeks29 (prophylactic) and four injections every other day12 (therapeutic). Because we use cholera toxin (CT) as an adjuvant, there is a possibility that tumor immunity is induced by CT and not the OVA mRNA nanoparticle. To rule out this possibility, we immunize mice with green fluorescent protein (GFP) mRNA nanoparticles as controls.

Immunization scheme for intranasal mRNA tumor vaccine.
(a) Prophylactic immunization. (b) Therapeutic immunization. (c) Dosing Scheme. (NP: mRNA nanoparticles, CT: Cholera Toxin, N: NALT, T: Turbinates). Additional details are provided in Materials and Methods.
Results
Prophylactic immunization with nasally administered mRNA vaccine
We tested intranasal immunization using a prophylactic tumor model, where mice were challenged with 4 × 105 E.G7-OVA cells injected into the left flank 7 days after the last immunization (Figure 1a). Mice intranasally immunized with OVA mRNA nanoparticles (mOVA-NP) demonstrated tumor inhibition (p < 0.01) and overall survival efficacy (p < 0.01) compared to mice immunized with GFP mRNA nanoparticles (mGFP-NP) or naked OVA mRNA (mOVA-N) (Figures 2a and b). The median tumor free and overall survival duration for the mOVA-NP group were 14.5 and 23 days, which were significantly longer compared to control groups (6 and 14 days for mGFP-NP group; 7 and 13 days for mOVA-N group, respectively). 2 out of 10 mice in the mOVA-NP group remained tumor free for the duration of the study (40 days). Notably, the tumor growth kinetics of mOVA-N group overlapped completely with negative control (mGFP-NP group, Figure 2c), indicating that intranasal administration with naked mRNA did not induce prophylactic tumor immunity. Hence, we concluded that intranasal vaccination with mRNA encapsulated in nanoparticle has an anti-tumor effect in the murine prophylactic E.G7-OVA tumor model.

Prophylactic immunization with OVA mRNA nanoparticles, but not GFP mRNA nanoparticles or naked OVA mRNA induces anti-tumor immunity.
Female C57Bl/6 mice were immunized as shown in Figure 1. 4 × 105 E.G7-OVA tumor cells were injected subcutaneously 7 days later. Mice were sacrificed once tumor volume reached 500 mm3. This experiment was conducted two times with similar results. Data from the second experiment are depicted. (a) Onset of palpable tumors. (b) Overall survival. (c) Tumor growth over 14 days. **p < 0.01.
Therapeutic immunization with nasally administered mRNA vaccine
We further evaluated the potential of intranasal mRNA tumor vaccination using a therapeutic tumor model, where mice were injected with 2 × 105 E.G7-OVA cells into the left flank 2 days before the first vaccine dose (Figure 1b). The number of tumor cells used in the therapeutic tumor model is half that used in the prophylactic model because of the increased stringency of a therapeutic protocol wherein tumor cells are implanted prior to start of immunizations. This provided a treatment window of 7–10 days to determine if the test vaccination regimen could have a potential therapeutic effect. The therapeutic immunization scheme (Figure 1b) was based on a similar protocol used for intranodal naked mRNA vaccination that entailed four immunizations performed every other day12. The median tumor free duration for mOVA-NP group (15 days) was statistically significant compared to the control mGFP-NP group (9 days, p < 0.01) but not mOVA-N group (11 days, p = 0.067) (Figure 3a). However, the median overall survival for mOVA-NP group (23.5 days) was significant when compared to both mGFP-NP group (15 days, p < 0.001) and mOVA-N group (17.5 days, p < 0.05). Two out of eight mice in the mOVA-NP group remained tumor free (Figure 3b) for the duration of the study (40 days).

Therapeutic immunization with OVA mRNA nanoparticles, but not GFP mRNA nanoparticles or naked OVA mRNA induces anti-tumor immunity.
Female C57Bl/6 mice were injected subcutaneously with 2 × 105 E.G7-OVA tumor cells. 2 days later mice were immunized as shown in Figure 1. Mice were sacrificed once tumor volume reached 500 mm3. This experiment was conducted two times with similar results. Data from the second experiment are depicted. (a) Onset of palpable tumors. (b) Overall survival. (c) Tumor growth over 16 days. (d) Specific growth rate of tumors over time. *p < 0.05; **p < 0.01; ***p < 0.001.
We observed that following tumor onset, it took a relatively long time for tumors to grow in the mOVA-NP group. The effect of immunization in controlling tumor growth rate was consistently observed in every animal in the mOVA-NP group (Figure 3c). To gain further insight, we analyzed specific tumor growth rates30 (Equation 2) of each tumor bearing animal every 48 hours for 16 days (Figure 3d). In the mGFP-NP and mOVA-N immunized mice, tumor volumes increased aggressively as soon as nascent tumors appeared. Specific growth rates were arrested rapidly from 0.9 day−1 (on day 2) to about 0.3 day−1 (day 6 and onwards) for reasons we speculated were related to tumor size (inefficient nutritional transport and onset of necrosis at later time points). In mOVA-NP immunized mice, specific growth rates did not progress at early time points and this translated into the observed growth delay (Figure 3c). Amongst the six tumor-bearing mice within the mOVA-NP group (two were tumor free), three showed negligible growth during the first 48 hours. Hence, we concluded that intranasal vaccination with mRNA encapsulated in nanoparticle format could also be effective for therapeutic tumor vaccination.
Tumor immunity requires mRNA to be delivered in nanoparticle but not in naked format
Because T cells are the major cell type involved in tumor clearance, we hypothesized that anti-tumor immunity observed in mOVA-NP treated mice (Figures 2 and 3) would correlate with the presence of OVA-specific T cells. Indeed, consistent with this hypothesis, we observed the presence of H-2Kb OVA tetramer+ CD8+ T cells in splenocytes isolated from mice immunized with mOVA-NP but not mGFP-NP or mOVA-N (Figure 4). Anti-tumor immunity was only observed in mice immunized with OVA mRNA delivered in nanoparticle format in both the prophylactic and therapeutic tumor model. These data suggests that the use of mRNA for intranasal vaccination applications will require delivery in nanoparticle format.

Induction of antigen-specific T cells following intranasal immunization with OVA mRNA nanoparticles.
(a) OVA-specific splenic CD8+ T cells stained with H-2Kb OVA tetramer. Mice (n = 2 per group) were immunized as shown in Figure 1 and described in Methods. Groups are mGFP-NP: mice immunized with GFP mRNA nanoparticles, mOVA-N: mice immunized with naked OVA mRNA and mOVA-NP: mice immunized with OVA mRNA nanoparticles. **p < 0.01. Results are presented as an average of 2 independent experiments. Representative data depicts analysis of cells harvested from mice immunized using the prophylactic model regimen with (b) mGFP-NP, (c) mOVA-N and (d) mOVA-NP. % H-2Kb OVA tetramer+ CD8+ T cells represents the percent of OVA tetramer positive cells within the CD8+ T cell population. Analysis of cells harvested from mice immunized using the therapeutic model regimen is shown in Supplementary Figure S3.
Discussion
In this proof-of-concept study, we demonstrate for the first time that intranasally administered mRNA encoding a tumor antigen can induce tumor immunity for the treatment of cancer. Our hypothesis is based on higher nasal transfection efficiencies and longer transgene expression kinetics achieved by mRNA nanoparticles as compared to mRNA delivered in the naked format. Mice treated with OVA mRNA encapsulated in nanoparticles, demonstrated delay in both tumor onset and overall survival compared to controls in prophylactic and therapeutic E.G7-OVA tumor model.
The overall survival and tumor onset of mOVA-NP group in the prophylactic model are clearly superior to mOVA-N group (Figure 2, Table 1). However, in therapeutic model the improvement is less distinct. When tumor growth kinetics between mOVA-N and mGFP-NP groups are compared, we observe that growth curves in the mOVA-N group completely overlapped with mGFP-NP group in the prophylactic model (Figure 2c), but a minor difference is seen in the therapeutic model (Figure 3c). This suggests that naked mRNA immunization had a slight effect in the latter. However, the difference in the latter is not statistically significant. The reason for this could be that the robust innate immune response mediated by cholera toxin facilitated the induction of an adaptive immune response following naked mRNA immunization in the nasal cavity. This corroborates data from another study demonstrating that nasally administered naked mRNA induces immune responses for the treatment of tuberculosis26.
In the therapeutic setting, we also observe that nascent tumors in all tumor-bearing mice treated with OVA mRNA nanoparticles do not proliferate aggressively. This could be attributed to immune response generated from intranasal immunization. Since tumor cells can escape immune surveillance31,32,33,34 through immune suppression35,36, altered expression of MHC class I37,38, as well as generation of immune escape tumor variants39,40, specific growth rates eventually caught up with that of control groups (Figures 3c and d). Lastly, through tetramer analysis we demonstrate that to induce anti-tumor immunity via intranasal route, it is necessary that mRNA is delivered in a nanoparticle.
Because intranasal delivery is a desirable route for vaccination, it has been extensively studied in the past decade. In particular, micro- and nano-particle delivery systems that encapsulate protein antigens or DNA that encode for antigens have been evaluated. However, the focus of intranasal vaccination has often been on the treatment of infectious diseases29,41,42,43,44,45,46,47. Nonetheless, a recent study has demonstrated that nanoparticles composed of modified γ-polyglutamic acid (γ-PGA) encapsulating full length OVA protein instilled intranasally induced anti-tumor immunity against melanoma48. In addition, a recent study also investigated the use of mannosylated chitosan-DNA (CS-DNA) nanoparticle vaccine for the prophylactic treatment of prostate carcinoma via the intranasal route. Anti-tumor response was only observed in the group that received targeted CS-DNA nanoparticles, but not in the group that received non-targeted nanoparticles. However, the therapeutic efficacy of the targeted CS-DNA nanoparticles was relatively similar to intramuscular vaccination using soluble antigen49.
Our results contribute to a relatively small number of studies published on mRNA nanoparticle mediated tumor vaccination where overall survival is one of the endpoints14,16,17,19. In addition, results from our current study also support the concept of nasal vaccination as an option for mRNA cancer immunotherapy. However, the therapeutic efficacy achieved in our current study is relatively moderate and we are uncertain how it compares with other administration routes or other established methods of mRNA vaccination. Therefore, future studies will focus on comparing this approach with other RNA-based methods and optimization of the current protocol to improve therapeutic efficacy.
For mRNA tumor vaccination to be clinically useful and broadly applicable, it is important that it is an off-the-shelf therapy that can be administered directly in vivo. In this report, we show that a convenient, non-invasive method can be used for direct in vivo administration of mRNA encoding tumor antigen, however it has to be delivered in nanoparticle format. This is an attractive prospect for the broad application of mRNA vaccines and reveals a major gap in the development of mRNA gene carriers for cancer immunotherapy.
Methods
Cloning of pGEM4Z/GFP/A64 and pGEM4Z/OVA/A64
The cDNA for green fluorescent protein (GFP) was derived from pEGFP-N1 (Clontech, Palo Alto, California) and inserted into pGEM4Z/A6450. Chicken ovalbumin cDNA in pUC18 was kindly provided by Dr. Barry T. Rouse, University of Tennessee, Knoxville. The 1.9 kb EcoR1 fragment containing the coding region and 3′ untranslated region was cloned into the EcoR1 site of pGEM4Z/A64 to generate plasmid pGEM4Z/OVA/A6451.
In vitro transcription of mRNA
Each plasmid of interest was digested with the restriction enzyme SpeI to linearize the DNA. After purification, DNA was used as template for in vitro transcription using T7 High Yield RNA Synthesis Kit (New England Biolabs, NEB) in the presence of anti-reverse cap analogue (ARCA, NEB) according to manufacturer's protocol. We routinely obtain 40–50 μg of OVA mRNA from a 20 μl reaction (1:3 GTP:ARCA mole ratio). In vitro transcribed (IVT) mRNA was purified with RNEasykit (Qiagen), quantified by spectrophotometry and analyzed by agarose gel electrophoresis to confirm the synthesis of full-length mRNA. GFP mRNA was labeled with Cy5 labeling kit (Mirusbio) according to manufacturer's protocol.
Nanoparticle formulation
mRNA nanoparticles were formulated (as previously described24) by adding 8 μl ethanol reagent (mRNA Transfection Reagent, Stemgent) to 10 μl of mRNA (0.2 μg/μl) suspended in Stemfect buffer under gentle vortexing for 10 seconds. The mixture was incubated at room temperature (RT) for 12 minutes under vacuum to completely remove ethanol. Size and zeta potential of nanoparticles were confirmed using NanoZS (Malvern) in both DI water (180 nm/+40 mV) and 10% FBS-supplemented media (300 nm, −12 mV), consistent with what we have previously reported24.
Ethics statement
In conducting the research described in this paper, the investigators adhered to the “Guide for the Care and Use of Laboratory Animals” as proposed by the committee on care of Laboratory Animal Resources Commission on Life Sciences, National Research Council. The facilities at the Duke University are fully accredited by the American Association for Accreditation of Laboratory Animal Care (AAALAC) and all studies were conducted using a protocol approved by the Duke University IACUC.
Intranasal vaccination
6 to 7-week old female C57Bl/6 mice were obtained from Jackson Laboratories. Intranasal immunization with mRNA encoding chicken ovalbumin (mOVA) or green fluorescent protein (mGFP) encapsulated in nanoparticles was performed according to Figure 1. Each nasal administration was done in 3 steps as detailed in Figure 1C. Mice were anesthesized with isofluorane in a gas chamber and queued for nasal administration. Each time a single mouse was taken out of the chamber, held in supine position, nasally administered with 15 μl of mRNA nanoparticles (3 μg) using a P20 pipette (fitted with a gel loading tip) and laid back inside the gas chamber in supine position. This procedure was repeated for the next animal in sequence. Consequently, each mouse was handled twice at an interval of approximately 5 minutes between each 15 μl dose for a total of 30 μl (6 μg) of mRNA nanoparticles. Mice were rested for 4 hours to allow gene expression to peak and subsequently administered with 1 μg cholera toxin (CT, List Biologicals) in 10 μl PBS. Mice were rested for another 2 hours to allow early immune response at the nasal site and subsequently administered with an additional 15 μl (3 μg) of mRNA nanoparticles. The procedure for naked OVA mRNA administration was identical to that used for OVA mRNA nanoparticles. In summary, each mouse received a total of 9 μg of OVA (or GFP) mRNA nanoparticles and 1 μg of CT per vaccination.
Tumor immunotherapy models
For prophylactic immunization, 4 × 105 E.G7-OVA tumor cells (in 100 μl PBS) were injected subcutaneously into the left flanks of immunized mice 7 days after the last immunization (Figure 1). For therapeutic immunization, 2 × 105 E.G7-OVA tumor cells (in 100 μl PBS) were injected subcutaneously into the left flanks of naïve mice 2 days before the first vaccine dose. Tumors were monitored every other day for tumor onset and measured with vernier calipers. Mice with tumors greater or equal to 500 mm3 were sacrificed. Tumor volume was calculated using Equation 1, where length is the longer of the 2 orthogonal measurements. Specific growth rate was calculated using Equation 230.


Where V2/V1 is the numerical ratio of tumor size measured from the same animal on respective days (D2 and D1).
Tetramer staining
Female C57Bl/6 mice were immunized using the prophylactic or therapeutic regimens as detailed above. 7 days after the last immunization, spleens were isolated and crushed through a 70 micron filter. Splenocytes were depleted of erythrocytes with ammonium chloride/Tris and re-suspended in PBS/10%FBS at a concentration of 107 cells/ml. Cells were blocked on ice with CD16/32 (Fc-block, BioLegend) for 15 minutes and subsequently stained with CD8-APC, isotype-PE antibodies (BioLegend) and PE-iTag-MHCI-OVA tetramer (Beckman Coulter) for 30 minutes on ice. Antibody staining was carried out per manufacturer's protocol. For tetramer staining, 2 μl of CD8-APC antibody and 5 μl of MHC class I H-2Kb OVA tetramer (amino acids 257–264, SIINFEKL) were added to 106 cells (in 100 μl) and incubated for 30 minutes at room temperature. Cells were washed, fixed with PBS/1% paraformaldehyde, data were acquired using flow cytometry (FACSCaliber, BD Biosciences) and analyzed using WinMDI 2.9 (freeware). Gating strategy for analysis of % H-2Kb OVA tetramer+ CD8+ T cells is described in Supplementary Figure S2.
Statistical analysis
For tumor studies, comparison between two groups was performed using the log-rank test (Mantel-Cox test). Additional comparisons between groups were done by determining the median survival for each group. Tumor growth curves over time were compared using two-way ANOVA with Bonferroni multiple comparison post-test. Statistical significance in tetramer staining comparing two groups was done using paired two-tailed Student's t test. A probability of less than 0.05 (p < 0.05) was considered statistically significant. Calculations were performed using GraphPad Prism.
References
Caruso, D. A. et al. Results of a phase 1 study utilizing monocyte-derived dendritic cells pulsed with tumor RNA in children and young adults with brain cancer. Neuro Oncol 6, 236–246 (2004).
Coosemans, A. et al. Immunological response after therapeutic vaccination with WT1 mRNA-loaded dendritic cells in end-stage endometrial carcinoma. Anticancer Res 30, 3709–3714 (2010).
Morse, M. A. et al. Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Invest 21, 341–349 (2003).
Rains, N., Cannan, R. J., Chen, W. & Stubbs, R. S. Development of a dendritic cell (DC)-based vaccine for patients with advanced colorectal cancer. Hepatogastroenterology 48, 347–351 (2001).
Steitz, J., Britten, C. M., Wolfel, T. & Tuting, T. Effective induction of anti-melanoma immunity following genetic vaccination with synthetic mRNA coding for the fusion protein EGFP.TRP2. Cancer Immunol Immunother 55, 246–253 (2006).
Su, Z. et al. Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J Immunol 174, 3798–3807 (2005).
Van Lint, S., Heirman, C., Thielemans, K. & Breckpot, K. mRNA: From a chemical blueprint for protein production to an off-the-shelf therapeutic. Hum Vaccin Immunother 9 (2013).
Nair, S., Boczkowski, D., Pruitt, S. & Urban, J. RNA in cancer vaccine therapy in Cancer Vaccines: From Research To Clinical Practice (ed Bot, A., Obrocea, M.& Marincola, F.) Ch. 16, 217–231 (Informa Healthcare, 2011).
Dannull, J. et al. Melanoma immunotherapy using mature DCs expressing the constitutive proteasome. J Clin Invest 123, 3135–3145, 10.1172/JCI67544 (2013).
Phua, K. K. et al. Whole Blood Cells Loaded with Messenger RNA as an Anti-Tumor Vaccine. Adv Healthc Mater, 10.1002/adhm.201300512 (2013).
Walch, B., Breinig, T., Schmitt, M. J. & Breinig, F. Delivery of functional DNA and messenger RNA to mammalian phagocytic cells by recombinant yeast. Gene Ther 19, 237–245, 10.1038/gt.2011.121 (2012).
Kreiter, S. et al. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res 70, 9031–9040, 10.1158/0008-5472.CAN-10-0699 (2010).
Van Lint, S. et al. Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res 72, 1661–1671, 10.1158/0008-5472.CAN-11-2957 (2012).
Hess, P. R., Boczkowski, D., Nair, S. K., Snyder, D. & Gilboa, E. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol Immunother 55, 672–683, 10.1007/s00262-005-0064-z (2006).
Hoerr, I., Obst, R., Rammensee, H. G. & Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol 30, 1–7 (2000).
Mockey, M. et al. mRNA-based cancer vaccine: prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther 14, 802–814, 10.1038/sj.cgt.7701072 (2007).
Perche, F. et al. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 7, 445–453, 10.1016/j.nano.2010.12.010 (2011).
Pollard, C. et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol Ther 21, 251–259, 10.1038/Mt.2012.202 (2013).
Zhou, W. Z. et al. RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum Gene Ther 10, 2719–2724, 10.1089/10430349950016762 (1999).
Kariko, K., Muramatsu, H., Keller, J. M. & Weissman, D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther 20, 948–953, 10.1038/mt.2012.7 (2012).
Su, X. F., Fricke, J., Kavanagh, D. G. & Irvine, D. J. In Vitro and in Vivo mRNA Delivery Using Lipid-Enveloped pH-Responsive Polymer Nanoparticles. Molecular Pharmaceutics 8, 774–787, 10.1021/Mp100390w (2011).
Zohra, F. T., Chowdhury, E. H., Nagaoka, M. & Akaike, T. Drastic effect of nanoapatite particles on liposome-mediated mRNA delivery to mammalian cells. Analytical Biochemistry 345, 164–166, 10.1016/J.Ab.2005.06.031 (2005).
Zou, S., Scarfo, K., Nantz, M. H. & Hecker, J. G. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int J Pharmaceut 389, 232–243, 10.1016/j.ijpharm.2010.01.019 (2010).
Phua, K. K., Leong, K. W. & Nair, S. K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J Control Release 166, 227–233, 10.1016/j.jconrel.2012.12.029 (2013).
Nochi, T. et al. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat Mater 9, 572–578, 10.1038/nmat2784 (2010).
Lorenzi, J. C. et al. Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol 10, 77, 10.1186/1472-6750-10-77 (2010).
Porgador, A., Staats, H. F., Faiola, B., Gilboa, E. & Palker, T. J. Intranasal immunization with CTL epitope peptides from HIV-1 or ovalbumin and the mucosal adjuvant cholera toxin induces peptide-specific CTLs and protection against tumor development in vivo. J Immunol 158, 834–841 (1997).
Cesta, M. F. Normal structure, function and histology of mucosa-associated lymphoid tissue. Toxicol Pathol 34, 599–608, 10.1080/01926230600865531 (2006).
Gwinn, W. M. et al. Effective induction of protective systemic immunity with nasally administered vaccines adjuvanted with IL-1. Vaccine 28, 6901–6914, 10.1016/j.vaccine.2010.08.006 (2010).
Mehrara, E., Forssell-Aronsson, E., Ahlman, H. & Bernhardt, P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res 67, 3970–3975, 10.1158/0008-5472.Can-06-3822 (2007).
Antonia, S. J., Extermann, M. & Flavell, R. A. Immunologic nonresponsiveness to tumors. Crit Rev Oncog 9, 35–41 (1998).
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. & Schreiber, R. D. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3, 991–998, 10.1038/ni1102-991 (2002).
Marincola, F. M., Jaffee, E. M., Hicklin, D. J. & Ferrone, S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74, 181–273 (2000).
Bodmer, W. F. et al. Tumor escape from immune response by variation in HLA expression and other mechanisms. Ann N Y Acad Sci 690, 42–49 (1993).
Baumgartner, J. et al. Melanoma induces immunosuppression by up-regulating FOXP3(+) regulatory T cells. J Surg Res 141, 72–77, 10.1016/j.jss.2007.03.053 (2007).
Rabinovich, G. A., Gabrilovich, D. & Sotomayor, E. M. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25, 267–296, 10.1146/annurev.immunol.25.022106.141609 (2007).
Aptsiauri, N. et al. Role of altered expression of HLA class I molecules in cancer progression. Adv Exp Med Biol 601, 123–131 (2007).
Restifo, N. P. et al. Molecular mechanisms used by tumors to escape immune recognition: immunogenetherapy and the cell biology of major histocompatibility complex class I. J Immunother Emphasis Tumor Immunol 14, 182–190 (1993).
Paschen, A. et al. Complete loss of HLA class I antigen expression on melanoma cells: a result of successive mutational events. Int J Cancer 103, 759–767, 10.1002/ijc.10906 (2003).
Sampson, J. H. et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 28, 4722–4729, 10.1200/JCO.2010.28.6963 (2010).
Fernandez, S., Cisney, E. D. & Ulrich, R. G. Enhancement of Serum and Mucosal Immune Responses to a Haemophilus influenzae Type B Vaccine by Intranasal Delivery. Clin Vaccine Immunol 20, 1690–1696, 10.1128/Cvi.00215-13 (2013).
Li, Y. H. et al. Enhanced immunogenicity of an anti-caries vaccine encoding a cell-surface protein antigen of Streptococcus mutans by intranasal DNA prime-protein boost immunization. J Gene Med 11, 1039–1047, 10.1002/Jgm.1386 (2009).
Mohan, T., Mitra, D. & Rao, D. N. Nasal delivery of PLG microparticle encapsulated defensin peptides adjuvanted gp41 antigen confers strong and long-lasting immunoprotective response against HIV-1. Immunol Res 58, 139–153, 10.1007/s12026-013-8428-5 (2014).
Nguyen, C. T., Kim, S. Y., Kim, M. S., Lee, S. E. & Rhee, J. H. Intranasal immunization with recombinant PspA fused with a flagellin enhances cross-protective immunity against Streptococcus pneumoniae infection in mice. Vaccine 29, 5731–5739, 10.1016/j.vaccine.2011.05.095 (2011).
Pontes, D. et al. Immune Response Elicited by DNA Vaccination Using Lactococcus lactis Is Modified by the Production of Surface Exposed Pathogenic Protein. Plos One 9, 10.1371/journal.pone.0084509 (2014).
Torrieri-Dramard, L. et al. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol Ther 19, 602–611, 10.1038/mt.2010.222 (2011).
Wu, Y. B. et al. Thermal-sensitive hydrogel as adjuvant-free vaccine delivery system for H5N1 intranasal immunization. Biomaterials 33, 2351–2360, 10.1016/j.biomaterials.2011.11.068 (2012).
Matsuo, K. et al. Intranasal immunization with poly(gamma-glutamic acid) nanoparticles entrapping antigenic proteins can induce potent tumor immunity. J Control Release 152, 310–316, 10.1016/j.jconrel.2011.03.009 (2011).
Yao, W., Peng, Y., Du, M., Luo, J. & Zong, L. Preventative vaccine-loaded mannosylated chitosan nanoparticles intended for nasal mucosal delivery enhance immune responses and potent tumor immunity. Mol Pharm 10, 2904–2914, 10.1021/mp4000053 (2013).
Nair, S. K. et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6, 1011–1017, 10.1038/79519 (2000).
Nair, S. et al. Injection of immature dendritic cells into adjuvant-treated skin obviates the need for ex vivo maturation. J Immunol 171, 6275–6282 (2003).
Acknowledgements
This work is supported by the Department of Defense (DoD) awards W81XWH-12-1-0260 (SK.N) and W81XWH-12-1-0261 (KW.L) and NUS-OGS by the National University of Singapore (KK.L.P).
Author information
Authors and Affiliations
Contributions
K.K.L.P. conceived the study. K.K.L.P. and H.F.S. designed the experiment. K.K.L.P. performed the experiment. K.K.L.P., S.K.N., K.W.L. analyzed, interpreted the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Data
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Phua, K., Staats, H., Leong, K. et al. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci Rep 4, 5128 (2014). https://doi.org/10.1038/srep05128
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05128
This article is cited by
-
Hyaluronic acid-antigens conjugates trigger potent immune response in both prophylactic and therapeutic immunization in a melanoma model
Drug Delivery and Translational Research (2023)
-
Messenger RNA translation enhancement by immune evasion proteins: a comparative study between EKB (vaccinia virus) and NS1 (influenza A virus)
Scientific Reports (2019)
-
mRNA vaccines — a new era in vaccinology
Nature Reviews Drug Discovery (2018)
-
Formulation, characterization and evaluation of mRNA-loaded dissolvable polymeric microneedles (RNApatch)
Scientific Reports (2018)
-
The changing shape of vaccination: improving immune responses through geometrical variations of a microdevice for immunization
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
