
Abstract
A broad swath of eukaryotic microbial biodiversity cannot be cultivated in the lab and is therefore inaccessible to conventional genome-wide comparative methods. One promising approach to study these lineages is single cell genomics (SCG), whereby an individual cell is captured from nature and genome data are produced from the amplified total DNA. Here we tested the efficacy of SCG to generate a draft genome assembly from a single sample, in this case a cell belonging to the broadly distributed MAST-4 uncultured marine stramenopiles. Using de novo gene prediction, we identified 6,996 protein-encoding genes in the MAST-4 genome. This genetic inventory was sufficient to place the cell within the ToL using multigene phylogenetics and provided preliminary insights into the complex evolutionary history of horizontal gene transfer (HGT) in the MAST-4 lineage.
Similar content being viewed by others


Introduction
Multigene phylogenetic analysis using cultivated microbial eukaryotes (protists) has provided an important backbone to the eukaryote tree of life (ToL)1 but has failed to address a fundamental problem with these taxa: sparse taxon sampling. This issue arises because many key lineages and in general most protist taxa cannot be successfully cultivated2,3 (at least long-term) or go undetected due to their small size or morphological simplicity (i.e., exist as cryptic species4). Therefore our understanding of the protist ToL is skewed by a preponderance of data from important parasites or easily cultivated free-living lineages. Another confounding issue is foreign gene acquisition either as result of plastid endosymbiosis (i.e., endosymbiotic gene transfer; EGT5,6) or horizontal gene transfer, HGT, from non-endosymbiotic sources7,8,9 that generates a reticulate history for many nuclear genes. A commonly used approach to address the massive scale of microbial eukaryotic biodiversity2 is DNA “barcoding” (e.g., using rDNA hypervariable regions10) to identify uncultured lineages. These data are however often insufficient to reliably reconstruct ToL phylogenetic relationships and do not address genome evolution. Another approach to studying the biology and evolution of uncultivated lineages is analysis of individual cells isolated using fluorescence-activated cell sorting (FACS) of natural samples followed by whole genome amplification (WGA) using multiple displacement amplification (MDA11,12,13,14,15,16). The pool of total DNA resulting from this process can be used to reconstruct the genomes of the host and associated symbionts, pathogens, or “food” DNA presumably present in cell vacuoles. This approach, termed single cell genomics (SCG) has been used to elucidate the phylogeny of individual cells and their biotic interactions13,14,15. Other applications that rely on MDA of single cells include targeted metagenomics, whereby marker genes are PCR-amplified from the DNA sample to decipher their distribution in ecosystems or larger fragments of DNA are assembled for analysis of gene content11,17.
Here we used SCG to generate the first draft genome assembly from a cell belonging to the broadly distributed group of MAST-4 uncultured marine stramenopiles18. MAST-4 cells are small-sized (ca. 2–5 μm diameter) protists that account for about 9% of heterotrophic flagellates in non-polar ocean waters19,20. Due to their high abundance, these cells are key bacterivores in marine environments, potentially controlling the growth and vertical distribution of bacterial species21 and playing important roles in nutrient re-mineralization22. Here we used a MAST-4 cell as a model to test SCG methods with uncultured taxa. The over-arching goal of our study was to assess the extent of genome completion that is possible when studying a single MDA sample. Analysis of the genome data using ab initio gene prediction identified 6,996 protein-encoding genes in the genome of the isolate. This represents >70% of the expected gene inventory of the MAST-4 lineage. Using these partial data we included the MAST-4 cell in the ToL using multigene phylogenetics and gained insights into its complex evolutionary history of horizontal gene transfer (HGT).
Results
Sample collection and preliminary analysis
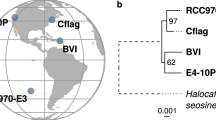
A water sample collected from Narragansett, Rhode Island, USA was sorted using FACS. Single heterotrophic cells <10 μm in size lacking chlorophyll autofluorescence were retained for MDA prior to rDNA identification and phylogenomic analysis. Analysis of 18S rDNA sequence showed that one was related to uncultured, heterotrophic stramenopiles identified in the English Channel and from Saanich Inlet in Vancouver, Canada (Fig. 1). High sequence identity of the stramenopile rDNA to taxa in the marine stramenopile group 4 (MAST-4; e.g., accessions RA010412.25, 14H3Te6O0, RA080215T.0778) identifies this cell as a member of this abundant, globally distributed member of the plankton that consumes bacteria and picophytoplankton18,22,23,24.

Analysis of protist SCG data.
Phylogenetic position of the Rhode Island MAST-4 single cell isolate used for genome sequencing. NCBI “gi” numbers are shown for each rDNA sequence. Known members of the MAST-4 clade24 are shown in red text. Bootstrap values shown above and below the branches are from RAxML and PhyML (in Italic text) analyses, respectively, using 1,000 iterations and the GTRGAMMA model of sequence evolution.
Genome assembly, gene prediction and search for contaminant DNA
A total of 6.62 Gbp of Illumina paired-end reads were generated from the MAST-4 MDA sample and assembled using SPAdes 2.425. This assembly had 123× average genome coverage and comprised 4,611 scaffolds with a total length of 16.93 Mbp (average scaffold length = 3,671 bp). The scaffold N50 was 14,108 bp and the maximum scaffold length was 111 Kbp (Table 1). We used the Core Eukaryotic Genes Mapping Approach (CEGMA26) to identify 159/458 conserved eukaryotic proteins in the MAST-4 SCG assembly. The genes encoding these proteins were used to predict27 6,996 proteins, of which 3,072 had a significant BLASTP hit (e-value ≤ 1e-10; Supplementary Fig. S1) with an alignment of at least 70% of the length of the protein to an existing sequence in our in-house peptide database (Supplementary Table S1). A vast majority of these top hits were to eukaryotic proteins. Relaxation of the BLASTP criteria to only the e-value cut-off and use of the NCBI “nr” database returned 4,645 hits, of which 4,091 (88%) of the top hits were to eukaryotes and 1,611 to stramenopiles (Supplementary Table S2).
We tested the possibility that some MAST-4 predicted proteins may have been contaminants derived from food sources, symbionts, or pathogens, as has been previously described in SCG work done with wild-caught, heterotrophic picozoan and Paulinella ovalis-like cells. These data contained significant bacterial, viral and phage DNA13,14,15,28 that assembled into contigs that were either very short, encoding a single gene or if larger (e.g., often >10 Kbp in length), encoded multiple prokaryote, viral, or phage genes. Many of these contigs had atypically high genome coverage due to MDA bias15,28. Given these data, we identified all prokaryote top hits to the MAST-4 predicted proteins. This analysis (Supplementary Table S3) turned up 294 scaffolds that contained a prokaryote top hit (total of 351 genes, 5% of the total). Of these, 119 scaffolds encoded a single prokaryote gene. All scaffolds with >1 prokaryote gene (232) also encoded genes of eukaryote origin, suggesting independent HGTs in these genome regions rather than contaminating prokaryote DNA. This is not surprising because protists have been shown to contain a large number of genes of prokaryote origin derived through HGT6,9,29. We however recognize that the MAST-4 assembly will likely contain some chimeric scaffolds (i.e., independently derived bacterial and eukaryotic genes that are artifactually assembled in one fragment), but it would be very surprising if all 232 scaffolds were chimeric. Finally, the 119 scaffolds that encode single prokaryotic genes comprise only 1.7% of the total gene set and had a minor impact on the overall phylogenetic analyses (see below).
The search for foreign eukaryote (i.e., prey or contamination) DNA in the assembly was repeated for MAST-4 proteins that had a BLASTP top hit to eukaryotes other than stramenopiles, alveolates, or rhizarians (the SAR clade). Although still a controversial issue, SAR taxa have been grouped together in phylogenies and likely share a sister group relationship30,31. This analysis turned up 13 scaffolds that encode ≥5 top hits to lineages such as metazoans, fungi and Viridiplantae (Supplementary Table S4). To investigate these results further, we generated a RAxML bootstrap tree for each predicted MAST-4 protein, resulting in 4,385 phylogenies. Manual analysis of trees derived from proteins encoded on the 13 scaffolds of interest did not identify any cases in which a single eukaryote source gave rise to the foreign genes. Most of these proteins were placed with SAR taxa in the RAxML trees (results not shown) indicating that the BLASTP result was not accurate. This suggests that eukaryote-derived food DNA likely does not exist in our MAST-4 assembly. We recognize however that proving clear instances of eukaryote-derived HGT (even from a single “food” lineage) is more difficult, in particular if the foreign sequences derive from SAR taxa. Intra-phylum HGT is difficult to identify without additional genomes to provide a more fine-grained history of stramenopile and SAR gene phylogeny.
Comparing results of the MAST-4 and control diatom MDA analyses
To explore the effectiveness of our approach to creating the MAST-4 assembly and predicted proteins, we generated three independent MDA samples (named A, B and C) from total DNA prepared from a unialgal culture of the diatom Thalassiosira pseudonana (CCMP1335). This species has a high-quality completed genome of size 34.5 Mbp that encodes 11,242 proteins32. Illumina paired-end data generated from the diatom MDA samples were assembled (Table 1) separately and when combined in one dataset and the encoded genes predicted (Table 2) as described above. Comparison of the MAST-4 SCG assembly with the assembly derived from the three T. pseudonana MDA samples indicates they are similar in terms of maximum scaffold and N50 lengths (Table 1).
The fraction of diatom reference proteins32 having at least 70% coverage with a BLASTP hit to our predicted proteins was 64%, 65%, 68% and 71%, respectively in the individual and combined diatom MDA-derived genome data (Supplementary Fig. S2A). In comparison, CEGMA recovered only 86.7% of the core proteins in the diatom reference assembly (that included organelle DNA) and the predicted proteins encompassed 73.40% of the reference diatom proteins with ≥70% coverage. Analysis of core CEGMA proteins within the predicted data from the MAST-4 SCG and diatom showed that ca. 60% and ≥90%, respectively, of the 458 genes were identified when compared to the Arabidopsis thaliana reference set (Supplementary Fig. S2B). When the alignment length of the MAST-4 and A. thaliana reference proteins was reduced to a minimum of 70%, then 326 (71%) core CEGMA proteins were identified in the MAST-4 data. This value climbed to 410 proteins (90%) when the alignment length was reduced to 50%. Analysis of the 248 ultra-conserved protein set in CEGMA showed that 89 complete proteins from this set was identified in the MAST-4 data. These results demonstrate that gene prediction from SCG data can detect a significant number of proteins (both complete and partial [i.e., the latter due to a fragmented assembly) in a microbial eukaryote without an available reference genome or transcriptome data.
The ca. 30% difference between the efficiency of recovery of complete proteins from the core set between the MAST-4 and diatom MDA samples (Supplemental Fig. S2A, S2B) likely reflects the fact that the diatom DNA was derived from a culture and therefore many copies of each chromosomal region were available for WGA. In contrast, the MAST-4 SCG data were derived from a single copy of the template DNA and some genome regions were apparently poorly sampled by MDA14,33 and missing from the final assembly. Consistent with this hypothesis, recent work with the highly reduced genome (1.6 Mbp) from the prokaryote endosymbiont Bartonella australis has demonstrated that the MDA procedure is significantly impacted by template DNA concentration34. These authors found that the level of MDA amplification bias was dependent on the quantity of template DNA, with greater amounts of starting material resulting in higher quality genome assemblies. Given these observations, pooling DNA from multiple cells derived from a single population of protists may result in better coverage of the target genome. In this case, however it would be critical to ascertain whether any of the single cells contain significant foreign DNA (e.g., prey bacteria, viruses) that could substantially weaken the combined cell assembly.
The hypothesis that data fragmentation and low coverage, rather than complete absence may provide an explanation for the low number of CEGMA proteins recovered from the MAST-4 data is impossible to address without a reference genome from this currently uncultivated lineage. However, consistent with this assertion is the finding that in comparison to the three T. pseudonana MDA samples that had significant representation of CEGMA proteins, the MAST-4 results provided similar assembly statistics and had a better average scaffold length (T. pseudonana Sample A = 1,176 bp, Sample B = 1,795 bp, Sample C = 1,875 bp, MAST-4 = 3,671 bp) suggesting these data are of comparable quality. We nonetheless recognize, as others have reported with prokaryotic MDA data35,36, that WGA methods do not provide complete coverage of a genome. Therefore missing data clearly contributes to the results of the CEGMA analysis with the MAST-4 cell. Genome reduction in the MAST-4 lineage may also play a role in explaining the CEGMA results. Gene loss is a common feature of many small-sized, mesophilic eukaryotes (e.g., Ostreococcus tauri, 12.6 Mbp37; Porphyridium purpureum, 19.7 Mbp9) and the heterotrophic MAST-4 cell is unlikely to be as gene-rich as a photosynthetic diatom.
Analysis of MAST-4 and diatom predicted proteins using KEGG
To gain preliminary insights into the functions encoded by MAST-4 predicted proteins, we mapped these sequences to the Kyoto Encyclopedia of Genes and Genomes (KEGG) categories. Given the incomplete assembly, we assumed in the KEGG analysis that missing proteins are randomly distributed across pathways. Therefore if a particular pathway is present in the MAST-4 cell, then it would be partially filled and if a pathway is absent in the KEGG analysis, then there is no reason to expect that this reflects an assembly artifact, but rather indicates true absence. Given this hypothesis, the KEGG analysis (Supplemental Fig. S3) demonstrates that many conserved pathways are present (e.g., TCA cycle), whereas others such as the urea cycle and photosynthesis are absent and presumably do not exist in the MAST-4 cell. For example, unlike the diatom no light harvesting chlorophyll complex proteins are present in the MAST-4 cell, as are missing nuclear encoded photosystem II precursors such as psbO and psbM. Genes present in MAST-4 that are missing in the diatom include a member of glycosyltransferase family 29 involved in galactose metabolism (protein 230; EC:3.2.1.23) and several enzymes involved in amino acid and nucleotide metabolism (Supplementary Fig. S3). Analysis of proteins involved in protein synthesis [e.g., ribosomal proteins, aminoacyl-tRNA biosynthesis (Supplementary Fig. S4)] and metabolic functions [e.g., purine metabolism, fatty acid metabolism (Supplementary Fig. S5)] shows that the majority of these pathway components are present in MAST-4.
Inference of the ToL
Phylogenomic analysis of individual MAST-4 proteins demonstrated the expected sister-group relationship to stramenopiles (e.g., diatoms and oomycetes) and minimal evidence of contaminating DNA (i.e., 94.8% of the trees summarized in Supplementary Fig. S6 show a eukaryotic affiliation for MAST-4 proteins). To test the usefulness of these data for inferring the eukaryotic ToL, we generated a concatenated alignment from a broad collection of completed genomes that incorporated the 458 CEGMA proteins (i.e., with some missing data from MAST-4 and other taxa). This alignment had a length of 25,145 amino acids and maximum likelihood analysis shows 100% RAxML bootstrap support for the phylogenetic affiliation of MAST-4 with other stramenopiles in a tree that provides robust bootstrap support for many other nodes (Fig. 2 and Supplementary Fig. S7). RAxML analysis of a reduced dataset of 159 CEGMA core eukaryote proteins (12,627 amino acids) that were complete in the MAST-4 assembly provided similar results (Fig. 2). Although the phylogenetic position of the MAST-4 lineage is well known based on rDNA comparisons (e.g., Fig. 1), the multi-gene analysis demonstrates the utility of SCG data for inferring the phylogenetic position of other uncultured eukaryotes that may be of unknown or uncertain affiliation. The phylogenomic output shows that a strong signal also exists for foreign gene acquisition in MAST-4 that conflicts with the topology of the CEGMA-based tree. These latter data reflect the expected HGT contribution to protist genomes5,6,7,8,9, but also may derive from alga-derived EGT that persists in MAST-4, potentially reflecting a photosynthetic past for this lineage38,39.

Analysis of proteins derived from MAST-4 SCG data.
Phylogenetic tree inferred from the concatenated alignment of the core 458 CEGMA proteins with the results of 100 bootstrap replicates (when ≥50%) shown at the branches. The numbers in Italics below the branches derive from a RAxML bootstrap analysis using a subset of 159 CEGMA proteins that were full-length in the MAST-4 SCG assembly. The complete tree is shown in Supplementary Fig. S7.
Using a protein coverage cut-off of ≥70% of the reference sequence in the database, 97 and 119 trees showed a sister group relationship between MAST-4 and green and red algal proteins, respectively (Supplementary Fig. S6). Two of these trees that incorporate GOS data are shown in Figure S8. Here a putative ion transporter protein (Supplementary Fig. S8A) is shared with and potentially derived from red algae in the MAST-4 cell as well as in photosynthetic haptophytes, rhizarians and the diatom Fragliariopsis cylindrus. Another example is a putative glutathione S-transferase protein (Supplementary Fig. S8B) shared with cyanobacteria, stramenopiles and rhizarians. More intriguing is the violaxanthin de-epoxidase (VDE) tree (Fig. 3a), in which all other taxa are photosynthetic eukaryotes. In algae and plants, plastid thylakoid localized VDE catalyzes the conversion of violaxanthin to zeaxanthin in the photo-protective xanthophyll cycle. The Mast-4 VDE-like protein appears to be green alga-derived, but in the non-photosynthetic stramenopile may function as a lipocalin, the broad functional group that gave rise to VDE40. This predicted protein 1451 lacks an N-terminal extension and its position in the genome contig suggests it is not the result of an assembly artifact or contamination (Fig. 3b); i.e., co-localized proteins in the contig [e.g., proteins 1447, 1448 and 1453 (Supplementary Fig. S9)] are of stramenopile or eukaryotic affiliation. Two other examples of putative algal-derived EGTs (a serine/threonine kinase domain containing protein and a myosin/kinesin motor domain, both shared with green algae) are shown in Supplementary Fig. S10.

Phylogeny of MAST-4 proteins.
(a) RAxML tree of violaxanthin de-epoxidase (VDE). The results of 100 RAxML and PhyML bootstrap replicates (when ≥50%) are above and below the branches, respectively and “gi” numbers are shown for taxa. (b) Coverage map and gene predictions for contig 104 in the MAST-4 SCG assembly that encodes the alga-derived VDE shown above.
Discussion
SCG is a rapidly developing field in medical and environmental science. In the latter area, virtually all work done until now has focused on prokaryotic taxa33 that have smaller, less complex genomes than eukaryotes. These microbes are more amenable to genome assembly when using MDA-derived sequence data. The initial work on marine protist SCG showed complex biotic interactions for taxa (e.g., presence of prey and pathogen DNA in the MDA sample13,14,15) that precluded a robust assembly of the host nuclear genome. Here we show that if a MDA sample derived from the natural environment is largely free of contaminating DNA, that it is possible to generate a useful, partial draft assembly from the cell. Applications of these SCG data include generating a gene inventory for the uncultivated taxon to study metabolic pathways and placing it in the ToL using multigene phylogenetics. With regard to inferring the ToL, our results demonstrate that the CEGMA proteins are well suited for this purpose even in the face of significant HGT in marine microbes. Regardless of how the MAST-4 trees are interpreted with respect to direction of gene transfer or the role of phylogenetic artifacts in explaining some of the topologies39, our data make it clear that the MAST-4 genome contains divergent phylogenetic histories. One of these histories (i.e., the CEGMA data) follows the expected trajectory of vertical inheritance, whereas the second involves a reticulate history of foreign gene sharing. This latter feature is typical of many photosynthetic algae (e.g., in Fig. 3a, Bigelowiella natans, Guillardia theta5,6,9) and as shown here characterizes the genome of a widely distributed plastid-lacking plankton.
In the future, generation of protist (particularly picoeukaryotic) marine metagenome data will potentially enable mapping of eukaryote-derived SCG proteins directly to natural environments. These data should provide the opportunity to elucidate the connection between the complex history of gene sharing via E/HGT and the global distribution of microbial eukaryotes5,6,7,9,40,41,42. Ultimately, it should be possible to map the global network of taxon-associated genes and gene families and to correlate these data to their evolutionary histories.
Methods
Cell collection and DNA preparation
A 500 mL sample of estuarine water was collected at low tide from the Pettaquamscutt River, Narragansett, Rhode Island, USA (41° 26′ 57.32″ N 71° 26′ 59.49″ W) on September 16th, 2009. The sample was kept in the dark and low temperature (−4°C) until processing, which was ≤ 6 hours thereafter at the Single Cell Genomics Center at the Bigelow Laboratory for Ocean Sciences, ME, USA. Environmental samples were pre-screened through a 70 μm mesh-size strainer (Becton Dickinson). A 3 mL subsample was incubated with the Lysotracker Green DND-26 (75 nmol L−1; Invitrogen) for 10 min in order to stain protist food vacuoles using pH-sensitive green fluorescence43. Target cells were identified and sorted using a MoFloTM (Beckman-Coulter) flow cytometer equipped with a 488 nm laser for excitation. Prior to target cell sorting, the cytometer was cleaned thoroughly with bleach. A 1% NaCl solution (0.2 μm filtered and UV treated) was used as sheath fluid44. Heterotrophic protist identification, single cell isolation and total DNA amplifications were carried out as described by Yoon and co-authors14. Two criteria were used for the heterotrophic cell sorting, the presence of Lysotracker fluorescence and <10 μm cell diameter. Cells were deposited into a 384-well PCR plate containing 0.6 mL of 1× TE buffer, centrifuged briefly and stored −80°C until further processing (plate no. AB108). Cells frozen in TE buffer were exposed to cold KOH for cell lysis and DNA denaturation45. Cell lysate genomic DNA was amplified using multiple displacement amplification (MDA46). The single cell amplified genomes were diluted 100-fold in sterile TE buffer, then screened by PCR and sequencing of conserved 18S rDNA. All sample 18S rDNA sequences were compared against RefSeq (http://www.ncbi.nih.nlm.gov/refseq) and their phylogenetic positions inferred based on a maximum likelihood tree with 100 bootstrap replications (RAxML Version 7.2.830). One MDA sample, AB108-RIc103, was identified as a member of the uncultured MAST-4 stramenopiles (Fig. 1). This sample was re-amplified using the REPLI-g Midi Kit (Qiagen) to generate sufficient DNA for downstream uses, following the manufacturer's instructions. The second MDA reaction was treated with the QIAqick PCR purification Kit (Qiagen). The MAST-4 library was prepared using the Nextera DNA sample prep kit (Illumina) according to the manufacturer's instructions using 50 ng of input DNA. Because the fragmentation was transposon-mediated, the average fragment size spanned the range 200–500 bp. Sequencing utilized the 500-cycle MiSeq Reagent Kit V2 (Illumina) in a 2 × 250 bp paired-end run.
For the diatom (T. pseudonana) sample, DNA was extracted from the algal culture using the Qiagen DNeasy Plant kit following the manufacturer's protocol. Three separate MDA reactions were done using the REPLI-g kit (Qiagen) according to the manufacturer's protocol starting with 50 ng of input diatom DNA in each reaction. Three sequencing libraries were prepared using the Nextera DNA Sample prep kit v2 and sequenced as described above. The three T. pseudonana libraries were multiplexed on one MiSeq sequencing run.
Single cell genome assembly
MDA to facilitate whole genome amplification (WGA) generates sufficient DNA from single cells for next generation sequencing [the recently described MALBAC47 procedure offers another promising approach]. MDA may however produce significant coverage bias, resulting in fragmented and incomplete assemblies35,36,48. Therefore, although it is clear that SCG may not provide complete genome assemblies, the challenge nonetheless is to maximize the quantity and quality of the data from uncultured taxa. Given these constraints, we applied a specialized assembler to the SCG data. Here SPAdes 2.425 was used because it demonstrates high performance when assembling bacterial single cell libraries (http://bioinf.spbau.ru/spades/). Initial assembly of the read library with default settings demonstrated that despite the maximum read length of 250 bp the median insert size was only 130 bp (with a standard deviation of 65 bp). In addition, the distribution of the library insert sizes varied widely and a significant amount of paired-end reads were of length 100–150 bp or shorter; many of the reads overlapped by 100–200 bp. In an attempt to improve the original data quality, we removed reads shorter than 150 bp in length, which increased the median insert size up to 200 bp but the resulting assembly was very fragmented due to loss of coverage. The unique iterative mode in SPAdes was used to recover the usable insert length and to preserve as much coverage as possible. The program was run using k-mer lengths of 21, 33, 55, 85, 95 and 127 in ‘careful’ mode. Using this approach, shorter k-mer lengths allowed us to keep the coverage, whereas longer k-mers exploited the usable part of insert size distribution to provide proper repeat resolution. Gene prediction began with derivation of 159 core eukaryote genes (CEGs) from our MAST-4 SCG assembly via CEGMA26. CEGMA is a computational method for building a highly reliable set of gene annotations in the absence of experimental (e.g., transcriptome) data. This approach relies on a defined a set of conserved protein families that occur in a wide range of eukaryotes and presents a mapping procedure that accurately identifies their exon-intron structures in a novel genome. This results in an initial set of reliable gene annotations in potentially any eukaryotic genome, even those in the draft stage. The CEGMA gene structures were then used to train the Augustus27 gene predictor prior to execution on the full genome assembly. Note that CEGMA only predicts genes for which a full-length homolog is found. Therefore, the fragmented nature of our assembly may have been the major reason for detecting only 159 (35%) of the core proteins. Interestingly, the set of proteins predicted by Augustus contained 243 CEGs (53%) with at least 60% BLASTP length similarity to a known Arabidopsis thaliana CEG.
KEGG ontology terms for the predicted MAST-4 proteins and for the known T. pseudonana proteome were obtained using the Automatic Annotation Server (http://www.genome.jp/tools/kaas/) with a (single/bidirectional) best hits method against the ortholog database. These terms were then used in the KEGG Mapper Pathway Reconstruction tool (http://www.genome.jp/kegg/tool/map_pathway.html) to annotate representative pathways.
Over-assembly of SCG data
As expected with MDA-derived genome data, we found a considerable fraction of over-assembly of diatom reads when compared to the T. pseudonana reference genome. The assemblies were larger than the reference genome (32.61 Mbp) by 38%, 25% and 35%, respectively for the three independent MDA samples and 47% for the combined data. It should however be noted that SCG assemblers such as IDBA-UD49 and SPAdes 2.425 were tested and optimized for bacterial datasets and there exists no dedicated SCG for larger, more complex eukaryotic genomes. Therefore, it was not our expectation that a perfect or near-perfect assembly would result from either the reference diatom or the MAST-4 genome. As a consequence, the set of predicted proteins we generated is also incomplete.
Phylogenomics
Phylogenomic analysis was performed as described previously5,9,50. Briefly, the MAST-4 predicted proteins were used in a BLASTP query against an in-house peptide database consisting of ca. 16.9 million sequences derived from RefSeq v.51 with the addition of sequenced eukaryote (e.g., Fungi, Metazoa, Viridiplantae and stramenopiles) taxa from the Joint Genome Institute (http://www.jgi.doe.gov) and 6-frame translated eukaryote EST sequences retrieved from NCBI dbEST (http://www.ncbi.nlm.nih.gov/dbEST). A taxonomically diverse set of target peptides were selected, aligned via MAFFT v641 and used for phylogenetic reconstruction under the PROTGAMMALG evolutionary model of RAxML v.7.2.83051. The resulting trees were sorted for patterns of monophyly using PhyloSort52. A multi-protein alignment of length 25,143 amino acids was produced by sampling our in-house database for homologs to the 458 CEGs. Each individual CEG family was aligned with its MAST-4 peptide homolog via MUSCLE53 and Gblocks54 was used to extract conserved sites from the alignment prior to concatenation and phylogenetic inference with RAxML (100 bootstraps, PROTGAMMALG model).
Additional information
Accession codes: The sequence data used to assemble the draft stramenopile MAST-4 single cell genome are available through NCBI BioProject PRJNA244411. The assembled genome, gene models, gene annotations, phylogenomic output, multigene alignments and other material are available at http://cyanophora.rutgers.edu/MAST4/.
References
Parfrey, L. W. et al. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst. Biol. 59, 518–533 (2010).
Pawlowski, J. et al. CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant and fungal kingdoms. PLoS Biol. 10, e1001419 (2012).
Guiry, M. D. How many species of algae are there? J. Phycol. 48, 1057–1063 (2012).
Boo, S. M. et al. Complex phylogeographic patterns in the freshwater alga Synura provide new insights on ubiquity versus endemism in microbial eukaryotes. Mol. Ecol. 19, 4328–4338 (2010).
Moustafa, A. et al. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324, 1724–1726 (2009).
Curtis, B. A. et al. Cryptophyte and chlorarachniophyte nuclear genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492, 59–65 (2012).
Keeling, P. J. & Palmer, J. D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 9, 605–618 (2008).
Chan, C. X. et al. Analysis of dinoflagellate genes reveals the remarkably complex evolutionary history of a microbial eukaryote. J. Phycol. 48, 1130–1142 (2012).
Bhattacharya, D. et al. Genome of the red alga Porphyridium purpureum. Nat. Commun. 4, 1941 (2013).
Behnke, A. et al. Depicting more accurate pictures of protistan community complexity using pyrosequencing of hypervariable SSU rRNA gene regions. Environ. Microbiol. 13, 340–349 (2011).
Cuvelier, M. L. et al. Targeted metagenomics and ecology of globally important uncultured eukaryotic phytoplankton. Proc. Natl. Acad. Sci. U.S.A. 107, 14679–14684 (2010).
Lepere, C. et al. Whole-genome amplification (WGA) of marine photosynthetic eukaryote populations. FEMS Microbiol. Ecol. 76, 513–523 (2011).
Worden, A. Z., Dupont, C. & Allen, A. E. Genomes of uncultured eukaryotes: sorting FACS from fiction. Genome Biol. 12, 117 (2011).
Yoon, H. S. et al. Single-cell genomics reveals organismal interactions in uncultivated marine protists. Science 332, 714–717 (2011).
Bhattacharya, D. et al. Single cell genome analysis supports a link between phagotrophy and primary plastid endosymbiosis. Sci. Rep. 2, 356 (2012).
Stepanauskas, R. Single cell genomics: an individual look at microbes. Curr. Opin. Microbiol. 15, 613–620 (2012).
Vaulot, D. et al. Metagenomes of the picoalga Bathycoccus from the Chile coastal upwelling. PLoS One 7, e39648 (2012).
Massana, R. et al. Phylogenetic and ecological analysis of novel marine stramenopiles. Appl. Environ. Microbiol. 70, 3528–3534 (2004).
Rodríguez-Martínez, R., Rocap, G., Logares, R., Romac, S. & Massana, R. Low evolutionary diversification in a widespread and abundant uncultured protist (MAST-4). Mol. Biol. Evol. 29, 1393–1406 (2012).
Logares, R. et al. Diversity patterns and activity of uncultured marine heterotrophic flagellates unveiled with pyrosequencing. ISME J. 6, 1823–1833 (2012).
Anderson, R., Wylezich, C., Glaubitz, S., Labrenz, M. & Jürgens, K. Impact of protist grazing on a key bacterial group for biogeochemical cycling in Baltic Sea pelagic oxic/anoxic interfaces. Environ. Microbiol. 15, 1580–1594 (2013).
Massana, R., Terrado, R., Forn, I., Lovejoy, C. & Pedrós-Alió, C. Distribution and abundance of uncultured heterotrophic flagellates in the world oceans. Environ. Microbiol. 8, 1515–1522 (2006).
Massana, R. et al. Phylogenetic and ecological analysis of novel marine stramenopiles. Appl. Environ. Microbiol. 70, 3528–3534 (2004).
Lin, Y. C. et al. Distribution patterns and phylogeny of marine stramenopiles in the North Pacific Ocean. Appl. Environ. Microbiol. 78, 3387–3399 (2012).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Stanke, M. & Morgenstern, B. Augustus: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Bhattacharya, D. et al. Identification of a marine cyanophage in a protist single cell metagenome assembly. J. Phycol. 49, 207–212 (2013).
Qiu, H. et al. Adaptation through horizontal gene transfer in the cryptoendolithic red alga Galdieria phlegrea. Curr. Biol. 23, R865–R866 (2013).
Hackett, J. D. et al. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of rhizaria with chromalveolates. Mol. Biol. Evol. 24, 1702–1713 (2007).
Burki, F., Okamoto, N., Pombert, J. F. & Keeling, P. J. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc. Biol. Sci. 279, 2246–2254 (2012).
Armbrust, E. V. et al. The genome of the diatom Thalassiosira pseudonana: ecology, evolution and metabolism. Science 306, 79–86 (2004).
Lloyd, K. G. et al. Predominant archaea in marine sediments degrade detrital proteins. Nature 496, 215–218 (2013).
Ellegaard, K. M., Klasson, L. & Andersson, S. G. Testing the reproducibility of multiple displacement amplification on genomes of clonal endosymbiont populations. PLoS One 8, e82319 (2013).
Rodrigue, S. et al. Whole genome amplification and de novo assembly of single bacterial cells. PLoS One 4, e6864 (2009).
Chitsaz, H. et al. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat. Biotechnol. 29, 915–921 (2011).
Palenik, B. et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. U.S.A. 104, 7705–7710 (2007).
Tyler, B. M. et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313, 1261–1266 (2006).
Stiller, J. W., Huang, J., Ding, Q., Tian, J. & Goodwillie, C. Are algal genes in nonphotosynthetic protists evidence of historical plastid endosymbioses? BMC Genomics 10, 484 (2009).
Frommolt, R. et al. Ancient recruitment by chromists of green algal genes encoding enzymes for carotenoid biosynthesis. Mol. Biol. Evol. 25, 2653–2567 (2008).
Archibald, J. M. et al. Lateral gene transfer and the evolution of plastid-targeted proteins in the secondary plastid-containing alga Bigelowiella natans. Proc. Natl. Acad. Sci. U.S.A. 100, 7678–7683 (2003).
Andersson, J. O. Gene transfer and diversification of microbial eukaryotes. Annu. Rev. Microbiol. 63, 177–93 (2009).
Rose, J. M., Caron, D. A., Sieracki, M. E. & Poulton, N. Counting heterotrophic nanoplanktonic protists in cultures and aquatic communities by flow cytometry. Aquat. Microbial Ecol. 34, 263–277 (2004).
Stepanauskas, R. & Sieracki, M. E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. U.S.A. 104, 9052–9057 (2007).
Raghunathan, A. et al. Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 71, 3342–3347 (2005).
Dean, F. B. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. U.S.A. 99, 5261–5266 (2002).
Zong, C., Lu, S., Chapman, A. R. & Xie, X. S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012).
Woyke, T. et al. Assembling the marine metagenome, one cell at a time. PLoS One 4, e5299 (2009).
Peng, Y. et al. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Price, D. C. et al. Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science 335, 843–847 (2012).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Moustafa, A. & Bhattacharya, D. PhyloSort: a user-friendly phylogenetic sorting tool and its application to estimating the cyanobacterial contribution to the nuclear genome of Chlamydomonas. BMC Evol. Biol. 8, 6 (2008).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577 (2007).
Acknowledgements
We thank members of the D.B. lab for helpful discussions, Kay Bidle for providing the diatom DNA and the School of Environmental and Biological Sciences and members of the Genome Cooperative at Rutgers for supporting this research. D.B. and H.S.Y. lab members are funded by the National Science Foundation and H.S.Y. is funded by a Korean RDA grant.
Author information
Authors and Affiliations
Contributions
E.C.Y. and H.S.Y. collected and prepared the single cell DNA that was processed at the Single Cell Genomics Center at the Bigelow Laboratory, ME, USA. G.C. was in charge of Illumina library preparation, single cell sequencing on the MiSeq instrument and was involved in helping to design the downstream bioinformatic analyses. D.C.P. processed the data and aided in genome assembly, gene prediction and phylogenomics. R.S.R. and A.K. generated the final genome assembly and gene models and R.S.R. did the phylogenomic analyses. D.B. generated the phylogenetic trees presented in the text. D.B., A.S. and R.S.R. designed the study. R.S.R., D.B. and D.C.P. wrote the paper. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Roy, R., Price, D., Schliep, A. et al. Single cell genome analysis of an uncultured heterotrophic stramenopile. Sci Rep 4, 4780 (2014). https://doi.org/10.1038/srep04780
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04780
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
