
Abstract
The last few years have seen the increasing development of new DNA targeting molecular tools and strategies for precise genome editing. However, opportunities subsist to either improve or expand the current toolbox and further broaden the scope of possible biotechnological applications. Here we report the discovery and the characterization of BurrH, a new modular DNA binding protein from Burkholderia rhizoxinica that is composed of highly polymorphic DNA targeting modules. We also engineered this scaffold to create a new class of designer nucleases that can be used to efficiently induce in vivo targeted mutagenesis and targeted gene insertion at a desired locus.
Similar content being viewed by others


Introduction
Exciting new possibilities in biotechnology and medicine have come with the ability to modulate cellular functions through the accurate and straightforward addition, removal, or exchange of DNA sequences1, alongside advances in and access to, genome sequencing and epigenetic information. To date, the meganuclease (MN)2,3, zinc finger (ZF)4,5, the recent CRISPR/Cas systems6,7 and the modular transcription activator-like effector (TALE)8,9,10 have proven to be versatile and robust architectures for generating molecular tools with customizable DNA specificity. The ease of assembly and the efficiency of such modular scaffolds for the specific targeting of DNA sequences have paved the way to broader and more diverse applications, especially in the field of synthetic biology which requires high-performing platforms with polyvalent characteristics. Surprisingly so far, although the number of fully sequenced bacterial genomes is approximately two thousand, functional TALE proteins have been identified and characterized only from two plant pathogens namely Xanthomonas spp.8 and Ralstonia solanacearum11. The constant release of new genomic data, notably from metagenomic samples, prompted us to search for new modular DNA binding proteins with potentially unique biochemical, biophysical and sequence/structural features.
In this study, using publicly available databases, we first carried out a bioinformatic search for putative DNA binding domains showing a few desired sequence/structural characteristics including: (i) the possibility to assemble the new putative DNA binding domain as modules, (ii) the possibility to design the specificity of the new putative DNA binding domain towards for individual nucleotides and as a novel feature (iii) a certain degree of polymorphism in the primary amino acid sequence (Fig. 1). We showed the identification of two new families of putative modular binding domains (M3BD) and the further characterization of one protein, thus demonstrating the potential to engineer this new scaffold to perform efficient genome editing in mammalian cells.

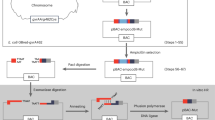
Strategy for identifying novel modular DNA targeting platforms.
Workflow used to identify new specific and modular DNA binding domains. Three principal features were used as guidelines for a bioinformatic search: modularity, polymorphism and specificity. Four families of databases were queried for different protein features: structure, sequence, localization, function. The outputs of the various databases were cross-referenced and manually inspected. The final filtered results provided the hits to be further characterized experimentally.
Results
Identification of new modular DNA targeting scaffolds
In order to identify new modular DNA targeting domain we firstly queried structural based databases such as SCOP12 and Cath13 together with Pfam14 (sequence based database based on Hidden Markov Model method) using the keywords “repeats +DNA”. This type of inquiring allowed us to extract 10 SCOP families and 27 CATH domains and 40 Pfam families. To refine our search we queried Pfam using the primary amino acid sequence of TAL effector PthXo115 retrieving the PFAM family PF03377, composed of 801 sequences, 12 architectures (sequence domains organization) and 45 species (such as Metazoa, Firmiticus, Proteobacteria and others). We next inspected the multiple sequence alignment (composed of the sequence motives of the PF03377 family), to be able to extract the complete sequence of the most promising hits (sequences composed of repeat motives having a certain degree of polymorphism). The best candidates were subsequently characterized for: prediction of secondary structure elements, protein localization (PSORT16, TargetP17) and prediction of function (Go18 and KEGG19). A few hits were then used as query for hmmsearch software (biosequence analysis software using hidden Markov model20) to retrieve possible hits in the metagenomic sequence repository.
Altogether, we identified two new families of putative modular DNA binding domains (M3BD for Modular Base-per-Base Binding Domain) (Table 1). The first family is composed of four protein fragments (containing 4 to 9 modules) from a metagenomic sample. The second family is composed of three proteins (containing 6, 20 and 27 modules) belonging to the proteome of Burkholderia rhizoxinica, a symbiotic bacterium living in the cytosol of Rhizopus microspores (Table 1 and Supplementary Fig. 1a, b, c). We have focused our further studies on one of the three proteins from Burkholderia rhizoxinica, identified under the accession number E5AV36 (UniProt21). This new scaffold protein will be further named BurrH, Fig. 2a.

Description of a new M3BD (Modular Base-per-Base Binding Domain) scaffolds from Burkholderia rhizoxinica.
(a) Sequence logo of the natural modules of AvrBs3 and BurrH as output by WEBLOGO. (b) Sequence and secondary structure prediction of the first module of BurrH. The sequence is indicated in single letter code. Cylinders represent helices (H). (c) Schematic layout of the organization of AvrBS3 (top) and BurrH (bottom). Colors represent the diversity of the modules.
A prediction of secondary structure elements (SSE) for the BurrH primary sequence revealed a high content of α-helices organized in a tight stretch of 20 supersecondary elements (modules) consisting of three α-helices linked by short loops (Fig. 2b). Globally, the predicted BurrH fold resembled the solenoid protein families such as tetratricopeptide (TPR), pentatricopeptide (PPR) and Sel1-like (SLR) repeat22. All these families share similar α-helical conformations but no conservation of primary sequence and superhelical topologies. Functionally they are mainly involved in protein-protein interactions but also in signal transduction pathways and polynucleotide recognition such as for TALE22.
The structural coordinate of a TALE (PDB code 3UGM, TPR fold15) was used to build a 3D model of the BurrH modules, Interestingly, we found notable differences between them (Fig. 2a, c). The BurrH modules revealed high polymorphism levels and key amino acid differences (Fig. 2b, c). Notably, the first position of a module was occupied by a large aromatic residue in BurrH (phenylalanine), whereas this was an aliphatic and more hydrophobic leucine in TALE (AvrBs3 TALE protein named AvrBs3-TALE8, was chosen as reference for comparison). Furthermore, clear inversions of polarity were revealed at several positions. In detail, positions 8 and 16, which surrounded the putative DNA binding loop, were occupied respectively by a lysine and an alanine in BurrH, but were inverted in AvrBs3-TALE (alanine in position 8 and lysine in position 16). Furthermore, the AvrBs3-TALE glutamate (position 20) and arginine (position 24) were substituted, respectively, by mainly positive residues (lysine, histidine but also glutamine) and negative residues (aspartate and glutamate) in BurrH (Fig. 2b). Finally, in BurrH, position 13 of the DNA binding loop was highly polymorphic, whereas the preceding position was consistently occupied by an asparagine (Fig 2b), suggesting a modular single amino acid base-per-base DNA recognition. Interestingly, two additional highly polymorphic cryptic modules that preserved the 8 K −16A feature were found at the N terminus of BurrH (Fig 2c). No detectable sequence identity could be found between these two BurrH cryptic modules and the two N-terminal TALE pseudo repeats, which were thought to be responsible for the specific recognition of a thymine at position 0 of the TALE DNA target8. Moreover, unlike all TALE proteins identified to date, the BurrH C-terminus lacked both a nuclear localization signal and a transcription activation domain (Fig. 2c)8.
Development of a BurrH-based nuclease
We assessed to what extent the BurrH scaffold could be engineered to target dsDNA in a sequence specific manner by creating an artificial nuclease. To determine a potential DNA target for the native BurrH binding core, we hypothesized that the biochemical and structural information from transcription activator-like effector (TALE) repeated units could be implemented for the development of the new BurrH scaffold23. Indeed TALEs were described with the specificity of DNA recognition resulting from two polymorphic amino acids located in positions 12 and 13 of a repeated unit (repeat variable di-residue, RVD). Access to the high resolution structure of TALEs bound to DNA showed that the amino acid at position 13 responsible for the specificity of recognition contacted the coding DNA strand base, whereas the amino acid at position 12 participated in the stabilization of the repeated units15,24. We thus designed a target for the BurrH protein using the following correspondence between the highly polymorphic position 13 of the BurrH modules and the DNA nucleotide: I = A, N = G, D = C, S = A, T = A, R = G. To create a designer nuclease (bn17421), we fused the catalytic domain of the FokI endonuclease to the C-terminal domain of the BurrH scaffold (Fig. 3a). To monitor the nuclease activity of this construct we took advantage of an existing yeast nuclease assay. This type of assay, widely used for the characterization of different classes of nucleases (e.g.: meganucleases, ZFNs and TALEN), is based on the restoring, through the single strand annealing (SSA) pathway, of a reporter gene (LacZ) upon cleavage by the nuclease25. The quantification of the β-galactosidase activity thus served as an indicator of DNA cleavage. In addition, the need to have two BurrH-based nuclease monomers because of the dimerization of the FokI catalytic domain prompted us to use particular homodimeric nuclease architecture. In this architecture, the targeted sequence (on the reporter plasmid) was composed of two duplicated sequences in inverse orientation and facing each other (separated by the so-called sequence spacer) on both DNA strands. Concerning spacer length, it had previously been shown for TALE nucleases (TALEN) that C-terminal truncated variants retaining 10–70 amino acids had the highest activity on spacer composed of 12–20 base pairs (preferably around 15 bp)26,27,28. We expected that a BurrH-based nuclease containing the complete 30 amino acid wild type C-terminal domain would exhibit similar behavior, so we designed our target to have a 15 bp spacer. To assess the specificity of cleavage, we additionally used a reporter plasmid where the targeted sequence was replaced by an irrelevant site (AvrBs3). The results we obtained in the yeast SSA assay clearly showed that this new construct led to an active and specific nuclease on its designed target (Fig. 3a, b and Supplementary Table 1), although the spacer length might not be optimal.

Engineering of the BurrH scaffold for the generation of target-specific nucleases in S. cerevisiae.
(a) Schematic representation of the BurrH-based nuclease bound to its designed target. All engineered BurrH-based nucleases contained an N-terminal nuclear localization sequence (NLS). (b) Activity of BurrH-based nuclease (bn17421) and AvrBs3-TALE nuclease (TALEN) on their respective homodimeric targets (spacer of 15 bp). Activity was measured using a Single Strand Annealing (SSA) based assay (Supplementary Fig. 2). Error bars denote s.d.; n ≥ 10. (c) Activity of BurrH-based nuclease (bn17421) 2 depending on the nucleotide in position −2, −1 or 0. A set of 20 homodimeric targets, separated by a DNA spacer of 15 bp and containing various combinations of the four bases in positions 0, −1 and −2 was created. Activity of BurrH-based nuclease was monitored as outlined in (b) individually on each target of the set. Each dataset contained at least three individual measurement points. Error bars denote s.d.; n ≥ 4. Control represents the mean value of experiments performed in absence of nuclease expressing vector.
In our effort to target the 18 nucleotide AvrBs3 sequence, the analysis of the BurrH sequence (20 modules) prompted us to engineer the DNA binding core principally by removing two modules in the middle or the natural BurrH array. We took into account both the finding related to the specificity of TALE nucleases and also the particular sequence features of the BurrH repeats. Indeed it had already been showed that TALE nuclease activity was more affected by mismatched bases at the 5′ of the target (targeted by the N-terminal repeats) than by mismatched bases at the 3′ of the target29, prompting the idea that N-terminal repeats guaranteed optimal activity/specificity and thus, by analogy, should be preserved in our engineered BurrH scaffold. At the same time, we observed that the C-terminal repeats of BurrH had unique sequence features that we speculated could be crucial to its activity. In the 19th and 20th repeats, position 8, prevalently occupied by a positively charged amino acid in the rest of the array, was substituted with a negatively charged amino acid (E). Starting with the 16th repeat, a few positions were mutated compared to the rest of the repeat array, namely positions 20, 24 and 25, substituted respectively by a lysine (a glutamine for the rest of the repeat array), a glutamate (substituted by an aspartic acid but also an alanine and a serine in the other modules) and an aromatic residue (a leucine in the rest of the array). We thus decided to remove the repeats 13th and 14th repeats in the middle of the array, as we hypothesized that the repeats at the two extremities could play a major role in the activity/specificity of BurrH derived nucleases, anyway we cannot exclude that a different choice of modules could create a BurrH-based nucleases with different features.
To benefit from the large body of information available on the specificity and activity of TALE nucleases while keeping the unique features of the primary sequence of the BurrH scaffold, we next evaluated the possibility of implementing only the four most often used RVDs from the AvrBs3-TALE8 into our new scaffold. Among these four RVDs from AvrBs3, three were already found in the native BurrH scaffold, so we kept the current association code (NI:A, NN:G and NG:T). In doing so, we replaced only the ND di-residue found in BurrH with the HD from AvrBs3 to target the cytosine nucleotide. We felt this choice was well supported, beacuse we noted that the ND di-amino acids had also been previously identified in known Xanthomonas TALE sequences and led to decreased activity, relative to HD, when incorporated into TALE-based engineered transcription activators23. In our final construct (bn18473), only the RVDs (positions 12 and 13) were exchanged, while the remaining primary sequence of the polymorphic modules was kept as in the wild type protein. Although we had previously found a strong activity with wild type scaffold, we also performed the complete activity screen to identify optimal spacer length for this engineered scaffold. The results we obtained clearly showed that the implementation of the four NI, HD, NN and NG RVDs led to an active nuclease and that, as hypothesized, optimal spacer sizes (13–15 and 23–25 bp) were similar to those of TALEN (Supplementary Fig. 3a).
To additionally get an insight in the capacity of BurrH based nuclease to accommodate mismatches in the targeted sequence (relative to the NI:A, HD:C, NN:G and NG:T code), we tested, using the yeast SSA assay, the bn18473 nuclease on a serie of 9 targets containing 2, 8, 14 or 16 mismatches (Supplementary Table 5). An engineered TALEN targeting the same sequence was also used as a reference for specificity (Supplementary Table 3). Although a larger in vivo data set would be desirable to precisely characterize the specificity of BurrH-based molecular tools. The analysis of this experiment showed similar behavior between TALE- and BurrH-based nucleases on targets containing mismatches (Supplementary Figure 3b).
Next, we investigated whether this new scaffold possessed any constraint on the base in position 0 of the target. It had previously been reported that both TALEs8 and the RipTALs11 required a specific base at this particular position to maximize their efficiency (thymine for TALEs and guanine for RipTALs). We therefore generated a set of targets containing various combinations of the four bases in positions 0, −1 and −2 (also to evaluate the potential impacts of the two cryptic modules). We demonstrated, using the bn17421 nuclease that DNA targeting with the BurrH scaffold was independent of the nature of the base at these positions (Fig. 3c).
In vivo activity of BurrH-based nucleases
To test the possibilities of this new molecular platform for cell engineering, we evaluated the ability of a BurrH-based nuclease to induce targeted mutagenesis in the 293H human cell line. For this purpose we chose to target a sequence in the 5′UTR of the housekeeping CAPNS1 gene, as this locus had previously been characterized30 and due to the availability of a TALE nuclease (TALEN) on the same sequence (Fig. 4a). We thus re-engineered the BurrH scaffold by changing only the RVDs to generate two designer nucleases composed of 20 (wild-type length, bn21604/21608) or 18 (engineered length, bn21603/21607) modules. Targeted mutagenesis, generated by the non-homologous end joining (NHEJ) repair pathway, at the double strand break (DSB) site, was estimated on the whole cell population three days post transfection. Thus we measured mutagenic event (small deletions and insertions, Indels) frequencies by performing a specific PCR of ~400 bp surrounding this CAPNS1 locus, followed by amplicon deep sequencing of genomic DNA. Remarkably, the BurrH-based nucleases induced efficient mutagenesis of up to 10% (Fig. 4a, b and Supplementary Fig. 4a, b) at the targeted locus, this being overall only moderately (~3-fold) less efficient than the corresponding TALEN (~30% Indels).

Characterization of BurrH-based nucleases in vivo (293H).
(a) Alignment of the WT genomic sequence and the most predominant events (deletions are indicated by dashes) induced by the BurrH-based nuclease (20 modules, bn21604/21608) at the CAPNS1 locus. Binding sites of the two half BurrH-based nuclease are underlined. (b) Size distribution of the deletion (blue bars) and insertion (red bars) events induced at the CAPNS1 locus by the BurrH-based nuclease composed of 20 modules. (c) Targeted gene insertion frequency determined at the CAPNS1 locus in the presence (+) or absence (-) of the nuclease. Error bars denote s.d.; n = 2.
In addition, to further assess the potential of the BurrH scaffold, we performed targeted gene insertion experiments at the same CAPNS1 locus. Thus we designed a plasmidic donor DNA that was composed of an heterologous fragment of 29 bp surrounded by two homology arms (~700 and ~500 bp) of the endogenous sequence located on both sides of the nuclease recognition site. Using a homologous recombination (HR) based strategy (Supplementary Fig. 5), we monitored specific insertions induced by the BurrH-based nuclease (20 modules). In our experimental setup, the cells were reseeded three days post transfection at a density of 10 cells/well in a 96-well plate, a strategy that was previously validated for designer nucleases30. We monitored targeted integration by performing, eighteen days post-transfection, for each well (288 wells in total), with two locus specific PCR amplifications, with one primer located within the heterologous insert of the donor DNA and the other on the genomic sequence outside of the homology arms. To evaluate the targeted gene insertion (TGI) frequency, we took into account the transfection efficiency (as monitored via GFP positive cells percentage) and plating efficiency (estimated at 30%). With this experimental setup, we found that the BurrH-based nuclease induced persistent events, with 8% of the cells, harboring targeted integration at the CAPNS1 locus (Fig. 4c). We further showed that this level of targeted gene insertion was similar to that observed for the TALEN (9%, Fig. 4c). As expected, no events were monitored in the absence of the nuclease, demonstrating that the double strand break created by the nuclease (BurrH-based or TALEN) was required for efficient homologous recombination.
In summary, the BurrH-based nucleases exhibited overall activity, for targeted mutagenesis as well as for targeted gene insertion, on par with that of currently available designer nucleases such as TALEN31,32. The long term persistence of events was a good indicator of the lack of toxicity induced by these nucleases and thus demonstrated the proficiency of the new BurrH scaffold for genome engineering applications.
Discussion
Using our methodology, we identified and subsequently engineered a new modular protein for the generation of nucleases with tailored DNA specificity. Very recently Horvath and colleagues also reported the identification of novel non-classical TAL effector homologues33. We expect the scope of applications to widen with the future development of such innovative modular architectures, presenting low degrees of identity within their DNA targeting modules. Indeed, it was recently reported that DNA tandem repeat motifs from a TALE scaffold were incompatible with lentiviral vector systems, due to the instability of their repeated core, unless a complete re-writing of the sequence was undertaken34,35. The rapid accumulation of new genomic data, including from metagenomic samples and from organisms living in extreme conditions (of pH as well as of temperature, salinity and light) will make it possible to identify new particular orthologs of already-described modular DNA targeting proteins. In summary, the results we presented here shed light on strategies for identifying and engineering novel modular DNA targeting platforms with unique sequence, biochemical, biophysical or structural properties. The distinct new features of the M3BD (Modular Base-per-Base Binding Domain) scaffolds we report here could further extend the current molecular toolboxes (e.g. repressors, transcription activators, nucleases, nickases) and expand the possibilities for genome editing, gene therapy and synthetic biology.
Methods
BurrH and TALE scaffolds and DNA targeting arrays
BurrH scaffolds and DNA targeting arrays were synthesized de novo (GeneCust) and subcloned sequentially in either yeast expression vectors or in mammalian expression vectors under the EF1α promoter. TALE scaffolds and DNA targeting arrays were obtained from Cellectis bioresearch (Paris). TALEN™ is a trademark owned by Cellectis bioresearch. All constructs contained a nuclear localization sequence (NLS). All relevant sequences and targets are presented in Supplementary Table 1–8.
Extrachromosomal SSA yeast assay
Nuclease containing yeast strains (mutant) were gridded using a colony gridder (QpixII, Genetix) on nylon filters placed on solid agar containing YP-glycerol, at a ~20 spots/cm2. A second layer, consisting of reporter-harboring (target) yeast strains, was gridded on the same filter. Filters were incubated overnight at 30°C to allow mating and then places and incubated for 2 days at 30°C on medium lacking leucine (for the mutant) and tryptophan (for the target) with glucose (2%) as the carbon source to allow selection of diploids. To induce the expression of the nuclease, filters were transferred onto YP-galactose-rich medium for 48 hours at 30°C or 37°C. Filters were finally placed on solid agarose medium containing 0.02% X-Gal in 0.5 M sodium phosphate buffer, pH 7.0, 0.1% SDS, 6% dimethyl formamide (DMF), 7 mM β-mercaptoethanol, 1% agarose and incubated at 30 or 37°C for up to 48 h to monitor nuclease activity, through the β-galactosidase activity. Filters were scanned and each spot was quantified using the median values of the pixels constituting the spot. We attribute the arbitrary values 0 and 1 to white and dark pixels, respectively. β-Galactosidase activity is directly associated with the efficiency of homologous recombination, thus with the cleavage efficiency of the nuclease.
Nuclease transfection
Human 293H cells (Life Technologies) were cultured at 37°C with 5% CO2 in DMEM complete medium supplemented with 2 mM l-glutamine, penicillin (100 IU/ml), streptomycin (100 μg/ml), amphotericin B (Fongizone: 0.25 μg/ml, Life Technologies,) and 10% FBS. 293H cells were seeded at 1.2 106 cells in 10 cm Petri dishes one day before transfection. Cell transfection was performed using the Lipofectamine 2000 reagent according to the manufacturer's instructions (Invitrogen). In brief, for targeted mutagenesis experiments, 2.5 μg of each of the two nuclease expression vector pairs and 10 ng of GFP expression vector (to monitor transfection efficiencies) were mixed with 0.3 ml of DMEM without FBS (5 μg final DNA amount). In another tube 25 μL of Lipofectamine were mixed with 0.3 ml of DMEM without FBS. After 5 minutes incubation, both DNA and Lipofectamine mixes were combined and incubated for 25 min at RT. The mixture was transferred to a Petri dish containing the 293H cells in 9 ml of complete and then cultured at 37°C under 5% CO2. Three days post-transfection, the cells were washed twice with phosphate-buffered saline (PBS), trypsinized, resuspended in 5 ml complete medium and the percentage of GFP positive cells was measured by flow cytometry (Guava EasyCyte) in order to monitor transfection efficacy.
For targeted gene insertion experiments, the same protocol was used excepted that 2.5 μg of each of the two nuclease expression vector pairs, 5 μg of circular donor DNA and 250 ng of GFP expression vector (to monitor transfection efficiencies) were mixed with 0.3 ml of DMEM without FBS (15 μg final DNA amount).
Targeted mutagenesis
Cells were pelleted by centrifugation and genomic DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer's instructions. PCR of the endogenous locus (393 bp) was performed using the oligonucleotide sequences presented in Supplementary Table 9 and purified using the AMPure kit (Invitrogen). Amplicons were further analyzed by deep sequencing using the 454 system (Roche).
Targeted gene insertion
Cells were re-seeded three days post-transfection in three 96 well plates at the density of 10 cells per well and cultured at 37°C for 15 more days in DMEM complete medium. The donor DNA, cloned in a plasmid, was composed of two homologous arms (717 bp and 538 bp) separated by 29 bp of an exogenous sequence. The frequency of Targeted Gene Insertion (TGI) events was monitored 18 days post transfection using TGI specific PCRs using the Herculase II Fusion kit (Agilent). The oligonucleotides used for the PCRs were designed to be located within the heterologous insert of the donor DNA for the first and on the genomic sequence outside the homology arms for the second. The sequences of these oligonucleotides are presented in Supplementary Table 9.
Computational methods
A survey of the different publicly available databases was performed by applying default parameters. The results were filtered and cross-referenced via manual inspection. Depending on the queried database different sources of inputs were used, for example sequences of known DNA binding proteins as well as super secondary structure elements reported as DNA binding motifs or simple short sequence motifs and also keywords. The databases principally used were: SCOP12, CATH13, Prosite36 Pfam14, Go18, KEGG19, InterPro37, TargetP1.117, PSORT16, NCBI (www.ncbi.nlm.nih.gov), HMMER20. Rosetta homology modeling38 (rosetta3.4) was used to build a model of the BurrH module using the structure of a TALE as template (PDB code 3UGM15).
References
Perez-Pinera, P., Ousterout, D. G. & Gersbach, C. A. Advances in targeted genome editing. Curr Opin Chem Biol 16, 268–277 (2012).
Silva, G. et al. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther 11, 11–27 (2012).
Pingoud, A. & Wende, W. Generation of novel nucleases with extended specificity by rational and combinatorial strategies. Chembiochem 12, 1495–1500 (2011).
Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 188, 773–782 (2011).
Sun, N., Abil, Z. & Zhao, H. Recent advances in targeted genome engineering in mammalian systems. Biotechnol J 7, 1074–1087 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Boch, J. et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512 (2009).
Bogdanove, A. J. & Voytas, D. F. TAL effectors: customizable proteins for DNA targeting. Science 333, 1843–1846 (2011).
Moscou, M. J. & Bogdanove, A. J. A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501 (2009).
de Lange, O. et al. Breaking the DNA-binding code of Ralstonia solanacearum TAL effectors provides new possibilities to generate plant resistance genes against bacterial wilt disease. New Phytol (2013).
Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 247, 536–540 (1995).
Sillitoe, I. et al. New functional families (FunFams) in CATH to improve the mapping of conserved functional sites to 3D structures. Nucleic Acids Res 41, D490–498 (2013).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res 40, D290–301 (2012).
Mak, A. N., Bradley, P., Cernadas, R. A., Bogdanove, A. J. & Stoddard, B. L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 335, 716–719 (2012).
Horton, P. et al. WoLF PSORT: protein localization predictor. Nucleic Acids Res 35, W585–587 (2007).
Emanuelsson, O., Brunak, S., von Heijne, G. & Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2, 953–971 (2007).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 27, 29–34 (1999).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–37 (2011).
The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res 37, D169–174 (2009).
Mittl, P. R. & Schneider-Brachert, W. Sel1-like repeat proteins in signal transduction. Cell Signal 19, 20–31 (2007).
Cong, L., Zhou, R., Kuo, Y. C., Cunniff, M. & Zhang, F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun 3, 968 (2012).
Stella, S. et al. Structure of the AvrBs3-DNA complex provides new insights into the initial thymine-recognition mechanism. Acta Crystallogr D Biol Crystallogr 69, 1707–1716 (2013).
Arnould, S. et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol 355, 443–458 (2006).
Miller, J. C. et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29, 143–148 (2011).
Christian, M. L. et al. Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS One 7, e45383 (2012).
Mussolino, C. et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res 39, 9283–9293 (2011).
Meckler, J. F. et al. Quantitative analysis of TALE-DNA interactions suggests polarity effects. Nucleic Acids Res 41, 4118–4128 (2013).
Daboussi, F. et al. Chromosomal context and epigenetic mechanisms control the efficacy of genome editing by rare-cutting designer endonucleases. Nucleic Acids Res 40, 6367–6379 (2012).
Kim, Y. et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31, 251–258 (2013).
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30, 460–465 (2012).
Schornack, S., Moscou, M. J., Ward, E. R. & Horvath, D. M. Engineering plant disease resistance based on TAL effectors. Annu Rev Phytopathol 51, 383–406 (2013).
Holkers, M. et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res 41, e63 (2012).
Yang, L. et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res (2013).
Sigrist, C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res 41, D344–347 (2013).
Hunter, S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40, D306–312 (2012).
Raman, S. et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 77 Suppl 9, 89–99 (2009).
Acknowledgements
We would like to thank Dan Voytas for critical reading of the manuscript. We acknowledge the contribution of the Cellectis Nuclease Production Platform.
Author information
Authors and Affiliations
Contributions
A.J., C.B., F.D. and P.D. conceived the study and designed experiments. A.J., C.B., G.D., V.G. and S.T. performed experiments. G.H.S., J.V. and M.B. provided conceptual advice. A.J., C.B., analyzed experiments. A.J., C.B. and P.D. wrote the manuscript with support from all authors.
Ethics declarations
Competing interests
All co-authors are Cellectis employees.
Electronic supplementary material
Supplementary Information
supplementary info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Juillerat, A., Bertonati, C., Dubois, G. et al. BurrH: a new modular DNA binding protein for genome engineering. Sci Rep 4, 3831 (2014). https://doi.org/10.1038/srep03831
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03831
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
