
Abstract
Glutaminase is a metabolic enzyme responsible for glutaminolysis, a process harnessed by cancer cells to feed their accelerated growth and proliferation. Among the glutaminase isoforms, human kidney-type glutaminase (KGA) is often upregulated in cancer and is thus touted as an attractive drug target. Here we report the active site inhibition mechanism of KGA through the crystal structure of the catalytic domain of KGA (cKGA) in complex with 6-diazo-5-oxo-L-norleucine (DON), a substrate analogue of glutamine. DON covalently binds with the active site Ser286 and interacts with residues such as Tyr249, Asn335, Glu381, Asn388, Tyr414, Tyr466 and Val484. The nucleophilic attack of Ser286 sidechain on DON releases the diazo group (N2) from the inhibitor and results in the formation of an enzyme-inhibitor complex. Mutational studies confirmed the key role of these residues in the activity of KGA. This study will be important in the development of KGA active site inhibitors for therapeutic interventions.
Similar content being viewed by others

Introduction
Besides aerobic glycolysis, or the Warburg effect, the involvement of glutaminolysis in cancers has received increasing attention as a potential avenue for the development of new therapeutic agents for cancer treatment1. Glutaminase controls the first step in the glutaminolysis pathway by converting glutamine (Gln) to glutamate (Glu), with subsequent enzymatic reactions generating aspartate, malate, pyruvate, citrate, alanine and lactate2. It has been widely accepted that cancer cells favour glutamine as a source of energy and this phenomenon has been observed in many cancers3,4. Thus, various studies postulate that inhibiting glutaminolysis by preventing the activity of this key enzyme would significantly hinder cancer cell growth and proliferation. Consequently, glutaminase has become an intriguing target for the development of drugs against human cancers5,6,7,8.
To date, three isoforms of human glutaminase have been identified: kidney-type (KGA/GLS1), the splice KGA variant (Glutaminase C or GAC) and liver-type (LGA/GLS2)9,10. Gao et al. recently showed that c-Myc stimulates KGA expression in P493 human B lymphoma and PC3 prostate cancer cells through direct suppression of miR23a and miR23b6. Similarly, activation of transforming growth factor beta (TGF-β) has been shown to stimulate KGA expression11. Importantly, the small GTPase, Rho, a pro-oncogenic molecule, can up-regulate KGA activity7 and we recently showed that KGA activity can be regulated by the Ras/Raf/Mek/Erk signalling cascade in response to growth factor stimulation8. Moreover, LGA was reported to be regulated by p53 in the energy producing-glutaminolysis pathway12,13. Collectively, these observations indicate that targeting KGA-mediated glutaminolysis could have high therapeutic implications with which to control cancer.
The glutamine analogue, 6-diazo-5-oxo-L-norlucine (DON), inhibits KGA and its isoforms by binding to the active site14. DON is a diazo compound and is known to interfere with both nucleotide and protein synthetic pathways where glutamine acts as a substrate15. However, DON lacks selectivity, as it also inhibits other glutamine-utilising enzymes such as the amidotransferases and glutamine synthetase16,17. The potential anti-cancer activity of DON was previously investigated in different animal models; however, concerns surrounding its toxicity prohibited its progression into clinical trials18,19. Similarly, a glutamate analogue, CK (L-2-amino-4-oxo-5- chloropentanoic acid), was also previously reported to act at the active site of KGA20 but was also not further explored. The recent renewed interest in cancer metabolism has raised the possibility that the optimization and systematic use of DON may have profound effects on human cancers.
The crystal structure of glutaminase from E.coli and B. subtilis in complex with DON has revealed the key role of a serine residue as a catalytic nucleophile21. We recently reported the crystal structure of the catalytic domain of kidney-type glutaminase (cKGA) in complex with L-glutamine and L-glutamate and proposed a catalytic mechanism for KGA8. The catalytic dyad of KGA consists of Ser286 and Lys289 (286-SCVK-289) and confirmed previous findings that the Ser286 acts as a catalytic nucleophile. In addition, we and others have reported the existence of a novel allosteric switch that governs the inhibition mechanism of KGA and GAC isoforms by BPTES8,22. Subsequently, Cassaga et al. proposed the activation mechanism of the GAC isoform by solving its crystal structure in complex with phosphate23.
To date, there have been no structural studies reported for any of the human glutaminase isoforms (KGA or GAC, LGA) in complex with their active site inhibitors. Here, we report the active site inhibition studies of KGA with several putative active site inhibitors as well as the crystal structure of cKGA in complex with DON. Similar to bacterial glutaminase, we show that DON forms a covalent bond with the active site Ser286 residue of KGA. Further, using site-directed mutagenesis, we validated the importance of various key residues involved in these interactions with DON. Taken together, these studies form the basis of a strategy to optimise KGA active site inhibitors for the improved and selective inhibition of this enzyme and may thus offer a first step toward the development of therapeutic intervention against glutamine-dependent cancers.
Results
Active site inhibition of cKGA by substrate analogue inhibitors
DON and azaserine are glutamine analogues that are known to inactivate several glutamine-utilising enzymes, such as glutaminase, amidotransferases and glutamine synthetase24. Besides, two additional glutamate analogues–glufosinate ammonium and L-methionine sulfoximine–were previously shown to inhibit the activity of glutamate-dependent enzymes25,26. We first conducted assays to identify which of these four inhibitors could best inhibit the activity of KGA (Figure 1). We found that DON inhibited cKGA significantly better than the other three inhibitors, with an IC50 of approximately 1 mM (Figure 1). Thus, DON was used in subsequent experiments to study the active site inhibition mechanism of KGA.

Comparison of the relative inhibition percentage of cKGA by selected glutamine/glutamate analogues at 1 mM concentration (IC50 of DON is approximately 1 mM).
The assays were performed in triplicates and values are mean ± SD of three independent experiments.
Structure of cKGA: DON complex
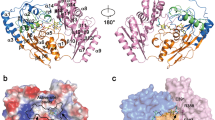
Molecular replacement method, using coordinates derived from apo cKGA (PDB code 3voy), was used to solve the cKGA: DON complex. The final model was refined with good stereochemical parameters (Figure 2A, Table 1). The complex structure of cKGA: DON shows that the inhibitor forms a covalent bond with the side-chain hydroxyl (OH) group of the catalytic nucleophile Ser286 (1.4 Å). This was confirmed by the continuous electron density from the active-site residue Ser286 with the inhibitor (Figure 2B). This Ser286 residue is conserved among all KGA isoforms. Using a one-to-one comparison based on the superposition of cKGA: DON and the apo-cKGA (rmsd of 0.3 Å for 315 Cα atoms), we observed no significant structural changes in cKGA upon its interaction with DON (Figure 3).

Structure of cKGA: DON complex.
(A) Ribbon representation of the cKGA: DON complex. Secondary structures α-helices and β-sheets are shown in dark green and magenta, respectively. DON is located in the active site cleft and is shown as a yellow stick. (B) Interactions made by DON in the catalytic pocket of cKGA. DON is shown in yellow and cKGA residues are shown in grey. Final 2Fo-Fc electron density map for DON and its covalent attachment to Ser286 of cKGA is shown. The map is contoured at σ level of 1.0. This figure and other structure-related figures reported in this paper were prepared using Pymol39.

Crystal structure of apo cKGA superimposed with cKGA: DON.
The active site residues of apo cKGA and cKGA: DON is shown in magenta and grey, respectively.
cKGA: DON interactions
The complex structure of cKGA: DON shows that the inhibitor binds in the catalytic pocket. The nucleophilic attack of the Ser286 side-chain on DON releases the diazo group (N2) from DON in the complex, thereby covalently connecting the enzyme with the 5-oxo-l-norleucine (ON) component of the inhibitor to form a stable cKGA-ON complex ( Figure 6). The bound inhibitor maintains several hydrogen bonding and hydrophobic interactions with Tyr249, Gln285, Ser286, Asn335, Glu381, Asn388, Tyr414, Tyr466 and Val484 of cKGA (Figure 2B). In particular, the carbonyl oxygen of the inhibitor is involved in two hydrogen-bonding contacts with the backbone amino (NH) group of Ser286 and Val484 and forms an oxyanione hole. Similarly, the α-carboxyl oxygens of the inhibitor are involved in two hydrogen-bonding contacts with the side-chains of Asn335 and Tyr414 of the enzyme. Moreover, the backbone NH group of the inhibitor is involved in a hydrogen bonding contact with the side-chains of Gln285 and Glu381 (Table 2). Besides these interactions, a hydrophobic cluster, formed by the side-chains of Tyr249, Tyr414 and Tyr466, further stabilises the interaction between cKGA and DON (Figure 2B). Notably, Tyr466 is located close to DON (3.2 Å) and also close to the catalytic Ser286 (2.7 Å), which is responsible for the proton transfer during catalysis. Previously, we proposed that Tyr466 played a key role in the catalytic mechanism along with the active site dyad (Ser286- CV-Lys289)8. The electrostatic surface potential of the cKGA: DON complex shows a predominantly positively charged active site region (Figure 4), with major contributions from the neighbouring residues (Lys299 and Lys491) of the active site region of cKGA.

Binding pocket of DON in cKGA.
(A) Shows the electrostatic surface potential of cKGA: DON complex.DON is shown as a yellow stick. (B) A close-up view of the electrostatic surface potential shows a positively charged pocket around DON. (C) The residues that interact with DON in the active site cleft are labeled.

Activities of wild-type cKGA compared with structure-guided cKGA alanine mutants.
Triplicate datasets were obtained for each of the cKGA constructs. Values are mean ± SD of three independent experiments.

Proposed active site inhibition mechanism of KGA.
The reaction mechanism progresses as follows: (i) Nucleophilic attack of Ser286 on DON; (ii) generation of covalent linkage between Ser286 and DON and the subsequent release of N2; (iii) formation of acetyl-inhibitor complex.
Mutational studies at the cKGA active site region
The complex structure of cKGA: DON revealed the key residues from cKGA that are essential for its interaction with DON. To validate the role of these key interacting residues of KGA in its enzymatic activity, we generated several alanine substitution mutants and measured their activities using a glutaminase assay. We found a significant reduction in the activity of cKGA mutants Tyr249A, Ser286A, Lys289A and Tyr466A as compared with the wild-type cKGA (Figure 5), which greatly affected substrate binding. Thus, our study confirms that the putative catalytic dyad Ser 289-CV-Lys289 and other residues, such as Tyr249 and Tyr466, play key roles in the activity of KGA.
Inhibition of KGA by DON
The cKGA: DON complex structure shows that DON is bound to the active site of KGA through covalent modification of the Ser286 residue. Based on our observations and previous literature, we propose a mechanism for the active site inhibition of KGA by DON (Figure 6). First, a hydroxyl proton from the active site Ser286 residue is donated to the diazo ketone of DON, resulting in the formation of the diazonium ion. Next, the nucleophilic attack of the Ser side-chain oxygen (Oγ) on the inhibitor releases N2 from the inhibitor. This is follwed by the generation of covalent linkage between Ser286 and the inhibitor, thus forming a stable enzyme-inhibitor complex. In our crystal strcture, we observed that the hydroxy oxygen of Ser286 is positioned about 1.4 Å from the δ carbon of DON. Based on this complex structure, it can be proposed that Lys289 of KGA acts as a general base to assist the catalytic Ser286 residue and this is further supported by a side-chain hydroxyl oxygen of Tyr 466. Moreover, the sequence conservation and structural homology between KGA and its homologues (e.g., glutaminase from E. coli and B. subtils) suggest a common mode of the active site inhibition mechanism21.
The superposition of cKGA: DON with YbgJ: DON (PDB code: 3BRM) gave a root mean square deviation (RMSD) of 1.3 Å for 285 Cα atoms. The cKGA has 36% sequence identity with its homologue YbgJ from B. subtilis. The active site residues of these two complex structures were well aligned, suggesting that human KGA may utilize similar catalytic mechanisms as its bacterial homolog (Figure 7A). Moreover, the superposition of cKGA: DON complex with cKGA: Glutamine (PDB code: 3VPO) gave a RMSD of 0.3 Å for 312 Cα atoms. Although the active site of both structures was identical, subtle differences in the position of bound DON in the active site can be observed (Figure 7B).

Structural comparisons.
(A) Superposition of cKGA: DON (grey) onto is homolog YbgJ from B. Subtilis (PDB code: 3BRM) (magenta). The key active site residues of cKGA are shown and the equivalent residues from YbgJ are shown in parenthesis. (B) Superposition of cKGA: DON (grey) and the cKGA: Glutamine (PDB code: 3VPO) (orange). The key active site region residues of both complexes of cKGA are shown. DON is shown in the final 2Fo-Fc electron density map (contoured at 1.0 σ).
Discussion
Recent studies have highlighted the importance of KGA as a promising molecular target for various human cancers5,6,7,8,22,23. Notably, small molecule anti-cancer inhibitors targeting KGA are in the early stage of clinical testing27; the success of these potential inhibitors, however, will require a clear understanding of the molecular mechanism of KGA inhibition. The current renewed interest in cancer metabolism raises the possibility that systematic treatment with DON, or another small molecule inhibitor, may be beneficial against human cancers.
In our studies, we found that DON significantly inhibits KGA at an approximate IC50 of 1 mM. Similarly, 2–10 mM DON can inhibit bacterial glutaminase (YbaS), although it acts as a covalent inhibitor21. A similar concentration range was reported for the covalent inhibition of human cystathionine gamma-lyase by DAG, which acts at an IC50 of 0.2 mM28. Moreover, several studies have reported the weak, irreversible and time-dependent inhibition mode of DON on glutaminase14,29,30,31. Whereas DON has been used widely to understand the functional role of KGA and other glutaminase isoforms, the lack of potency and selectivity of the compound has hindered its use in therapeutics. Hence, it is necessary to optimise the specificity of DON towards KGA for it to be a relevant option. The structure of the human cKGA: DON complex provides insight into the mode of binding and its activity, which will be important for the optimization, design and synthesis of a new series of DON analogues. In addition, BPTES and compound 968 (a dibenzophenanthridine) were recently identified as allosteric inhibitors of KGA and shown to block cancer cell growth and proliferation5,7,8. We recently elucidated the mechanism of allosteric inhibition of KGA by BPTES, showing that BPTES binds to KGA at the dimeric interface near the active site and destabilises the activity of enzyme through a drastic conformational change in a key loop (Glu312-Pro329)8. We further showed that alanine substitutions in this allosteric loop caused a reduction in the enzymatic activity as compared with wild-type, suggesting that the BPTES binding loop residues are essential for conferring KGA activity8. Furthermore, it was recently proposed that lysine (Lys 316) acetylation in the key loop is important for the regulation and activation of glutaminase32.
Glutaminase has been found to be elevated in lymphoma, prostate, glioblastoma, breast and kidney cancer cells5,6,7,8. It was recently reported that Myc stimulates glutaminase (GAC) expression, which directly increases the levels of glutamine metabolism in PC3 and P493 cell lines as compared with normal cells6. Notably, in response to Myc, lymphoma cells have shown about a 10-fold increase in the expression of glutaminase. Similarly it was also shown that Rho GTPase enhances glutaminase (GAC) expression in breast and transformed fibroblast cancer cells7. In addition to increasing protein expression levels described in these studies, we have recently shown that Ras/Mek kinase could activate the intrinsic activity of KGA in transformed cancer cells through phosphorylation without altering protein expression levels8. When used at 50 μM, nearly all of the KGA activity from in cells overexpressing KGA could be inhibited by BPTES, without causing much cytotoxicity to the cells8. Collectively, these observations suggest that glutaminase activity could be differentially regulated in cancer cells through alternate mechanisms and the findings point to the urgent need to design a new generation of hybrid inhibitors–with high potency and less toxicity–that selectively inhibit KGA in cancer cells.
To develop better inhibitors that specifically and effectively target KGA, we propose a strategy that targets both the allosteric site and the active site of KGA. This potential new inhibitor could combine optimised derivatives of both BPTES and DON connected via a flexible linker and acts to inhibit KGA by the mechanisms of each inhibitor. This approach is feasible, as the binding sites of these two inhibitors are approximately 8 Å away, as shown in our inhibition model generated using the independent BPTES (allosteric) and DON (active site) (Figure 8). This hybrid inhibitor has the advantage of retaining the specificity of BPTES and simultaneously targeting the active site and allosteric site. Besides, this strategy of inhibition could help to improve the pharmacophore properties and lead to the development of a group of next-generation inhibitors of glutaminase (KGA) with greater potency against KGA, with reduced toxic effects.

A model for a proposed hybrid inhibition of KGA combines the actions of BPTES and DON.
This model was prepared by superimposing the crystal structure of BPTES: cKGA and DON: cKGA. BPTES binds to the allosteric site, whereas DON binds to the active site region. These two sites are separated by approximately 8 Å, indicating the feasibility of this approach.
Methods
Expression and purification of cKGA
Human cKGA was purified following the procedures described previously8. Briefly, cKGA (Ile221-Leu533) was cloned into Pet-28(b) vector and Escherichia coli BL21 (DE3)-RIL-Codon plus cells were used to express the protein. The protein was purified using Ni-NTA affinity columns followed by gel filtration chromatography. The buffer conditions were 20 mM Na-Hepes pH 7.5, 200 mM NaCl, 10% glycerol and 2 mM DTT. DON (6-diazo-5-oxo-L-norleucine, NSC 7365), azaserine (O-diazoacetyl-L-serine, NSC 742), Glufosinate Ammonium and L-Methionine Sulfoximine were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Glutaminase assay
A glutaminase assay was performed using the two-step assay described previously8,33. Briefly, 10 μL of wild-type cKGA or cKGA harbouring alanine mutations was incubated at 37°C for 10 min with 10 μL of assay mix consisting of 20 mM glutamine, 50 mM Tris·acetate (pH 8.6), 100 mM phosphate and 0.2 mM EDTA. The reaction was quenched with the addition of 2 μL of 3 M HCl. Subsequently, the reaction mixture was incubated for 30 min at room temperature with 200 μL of the second assay mix (2.2 U glutamate dehydrogenase, 80 mM Tris·acetate (pH 9.4), 200 mM hydrazine, 0.25 mM ADP and 2 mM nicotinamide adenine dinucleotide). The absorbance was read at 340 nm using a microplate reader (SpectraMax 340; Molecular Devices, Sunnyvale, CA, USA).
Crystallization and data collection
Crystallization screening was carried out by hanging drop vapour diffusion at 22°C. Prior to crystallization, the complex was prepared by incubating approximately 20 mg/mL of cKGA with 10 mM DON (approximately 1:10 molar ratio) for 30 min at 4°C. The crystallization drop consisted of 1 μl cKGA: DON complex and 1 μl of the crystallization solution from the reservoir well. The cKGA: DON crystals grew after 2 days in the presence of a solution comprising 0.1 M Bis-Tris propane (pH 7.2) and 1.8 M LiSO4. For the diffraction studies, crystals were cryo-protected with reservoir solution supplemented with 15% glycerol as a cryo-protectant and flash-frozen in liquid nitrogen. Diffraction data were collected at the synchrotron beam line 13B1 (wavelength 1.000 Å) at the National Synchrotron Radiation Research Centre (NSRRC, Taiwan). Data sets were processed and scaled using HKL200034. The crystallographic statistics are provided in Table 1.
Structure solution and refinement
The structure of cKGA-DON complex was determined by molecular replacement with the program MolRep35 using the coordinates of apo cKGA as the search model (Protein data bank, 3VOY)8. One molecule of cKGA: DON complex was observed in the asymmetric unit and the initial R-factor was 39%. The model was examined and built in COOT36 and subsequent refinement was carried out with Phenix-refine37. Fitting of the inhibitor and refinements were performed in COOT and Phenix programs, respectively. The final structure was refined up to 2.3 Å resolution with an R-factor of 0.19 (Rfree 0.21) (Table 1). The overall geometry of the final model was analysed by Ramachandran plot with the program PROCHECK38.
Site-directed mutagenesis of cKGA
To create all structure-guided mutants of cKGA, we performed site-directed mutagenesis using a kit from Kapa Biosystems, Inc. (Woburn, MA, USA). cKGA cloned into the PET-28(b) vector was used as a template for mutagenesis. Key residues (Tyr249, Ser286, Lys289 and Tyr466) of cKGA were mutated to Ala as a single-point substitution. All mutations were verified by DNA sequencing. Plasmids containing the point mutations were transformed into the E. coli BL21 (DE3) RIL strain and the mutant cKGA proteins were over-expressed and purified following the same procedure as the wild-type cKGA.
Protein data bank accession code
Coordinates and structure factors of cKGA: DON complex have been deposited with RCSB Protein Data Bank under access code 407D.
References
Bensinger, S. J. & Christofk, H. R. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol 23, 352–361 (2012).
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G. & Thompson, C. B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7, 11–20 (2008).
Rajagopalan, K. N. & DeBerardinis, R. J. Role of glutamine in cancer: therapeutic and imaging implications. J Nucl Med 52, 1005–1008 (2011).
DeBerardinis, R. J. & Cheng, T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29, 313–324 (2010).
Seltzer, M. J. et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70, 8981–8987 (2010).
Gao, P. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009).
Wang, J. B. et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219 (2010).
Thangavelu, K. et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A 109, 7705–7710 (2012).
Aledo, J. C., Gomez-Fabre, P. M., Olalla, L. & Marquez, J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome 11, 1107–1110 (2000).
Curthoys, N. P. & Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr 15, 133–159 (1995).
Andratsch, M. et al. TGF-beta signaling and its effect on glutaminase expression in LLC-PK1-FBPase+ cells. Am J Physiol Renal Physiol 293, F846–853 (2007).
Hu, W. et al. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A 107, 7455–7460 (2010).
Suzuki, S. et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 107, 7461–7466 (2010).
Shapiro, R. A., Clark, V. M. & Curthoys, N. P. Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site. J Biol Chem 254, 2835–2838 (1979).
Kisner, D. L., Catane, R. & Muggia, F. M. The rediscovery of DON (6-diazo-5-oxo-L-norleucine). Recent Results Cancer Res 74, 258–263 (1980).
Ortlund, E., Lacount, M. W., Lewinski, K. & Lebioda, L. Reactions of Pseudomonas 7A glutaminase-asparaginase with diazo analogues of glutamine and asparagine result in unexpected covalent inhibitions and suggests an unusual catalytic triad Thr-Tyr-Glu. Biochemistry 39, 1199–1204 (2000).
Pinkus, L. M. Glutamine binding sites. Methods Enzymol 46, 414–427 (1977).
Ahluwalia, G. S., Grem, J. L., Hao, Z. & Cooney, D. A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther 46, 243–271 (1990).
Ovejera, A. A., Houchens, D. P., Catane, R., Sheridan, M. A. & Muggia, F. M. Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res 39, 3220–3224 (1979).
Shapiro, R. A., Clark, V. M. & Curthoys, N. P. Covalent interaction of L-2-amino-4-oxo-5-chloropentanoic acid with rat renal phosphate-dependent glutaminase. Evidence for a specific glutamate binding site and of subunit heterogeneity. J Biol Chem 253, 7086–7090 (1978).
Brown, G. et al. Functional and structural characterization of four glutaminases from Escherichia coli and Bacillus subtilis. Biochemistry 47, 5724–5735 (2008).
DeLaBarre, B. et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770 (2011).
Cassago, A. et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 109, 1092–1097 (2012).
Barclay, R. K. & Phillipps, M. A. Effects of 6-diazo-5-oxol-norleucine and other tumor inhibitors on the biosynthesis of nicotinamide adenine dinucleotide in mice. Cancer Res 26, 282–286 (1966).
Gill, H. S. & Eisenberg, D. The crystal structure of phosphinothricin in the active site of glutamine synthetase illuminates the mechanism of enzymatic inhibition. Biochemistry 40, 1903–1912 (2001).
Liaw, S. H. & Eisenberg, D. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 33, 675–681 (1994).
Garber, K. Oncology's energetic pipeline. Nat Biotechnol 28, 888–891 (2010).
Sun, Q. et al. Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J Biol Chem 284, 3076–3085 (2009).
Shijie, J. et al. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med 217, 87–92 (2009).
Thomas, A. G. et al. Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun 438, 243–248 (2013).
Hartman, S. C. & McGrath, T. F. Glutaminase A of escherichia coli. Reactions with the substrate analogue, 6-diazo-5-oxonorleucine. J Biol Chem 248, 8506–8510 (1973).
Ferreira, A. P. et al. Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem 288, 28009–28020 (2013).
Kenny, J. et al. Bacterial expression, purification and characterization of rat kidney-type mitochondrial glutaminase. Protein Expr Purif 31, 140–148 (2003).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A 276, 307–326 (1997).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66, 22–25 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221 (2010).
Laskowski, R. A., Macarthur, M. W., Moss, D. S. & Thornton, J. M. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. Journal of Applied Crystallography 26, 283–291 (1993).
DeLano, W. L. & Lam, J. W. PyMOL: A communications tool for computational models. Abstracts of Papers of the American Chemical Society 230, U1371–U1372 (2005).
Acknowledgements
We are grateful to the Biomedical Research Council of Singapore (BMRC), A*STAR (R154000461305) for the funding support. The X-ray diffraction data for this study were collected at the synchrotron beamline 13B1 at the National Synchrotron Radiation Research Centre (NSRRC, Taiwan). KT is a graduate scholar in receipt of a research scholarship from the National University of Singapore.
Author information
Authors and Affiliations
Contributions
J.S. conceived of the study. K.T. carried out crystallographic experiments. K.T. and J.S. analyzed the data. K.T. and Q.Y.C. performed mutation and kinetic experiments. L.B.C. was involved in the analysis. K.T. and J.S. wrote the manuscript with input from all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Thangavelu, K., Chong, Q., Low, B. et al. Structural Basis for the Active Site Inhibition Mechanism of Human Kidney-Type Glutaminase (KGA). Sci Rep 4, 3827 (2014). https://doi.org/10.1038/srep03827
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03827
This article is cited by
-
Glutamine increases stability of TPH1 mRNA via p38 mitogen-activated kinase in mouse mastocytoma cells
Molecular Biology Reports (2023)
-
Pharmacological inhibition of tumor anabolism and host catabolism as a cancer therapy
Scientific Reports (2021)
-
Molecular modeling and LC–MS-based metabolomics of a glutamine-valproic acid (Gln-VPA) derivative on HeLa cells
Molecular Diversity (2021)
-
Targeting GLS1 to cancer therapy through glutamine metabolism
Clinical and Translational Oncology (2021)
-
A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
