
Abstract
Idiopathic congenital nystagmus (ICN) consists of involuntary and periodic ocular motility, often with seriously reduced visual acuity. To identify the genetic defects associated with X-linked ICN, we performed PCR-based DNA direct sequencing of two candidate genes, FRMD7 and GPR143, in four families. Mutation analysis led to identification of three novel mutations, p.S260R, p.Q487X and p.V549Y fsX554, in FRMD7 in three of the recruited families. Results from structural modeling indicated that the p.S260R may potentially disrupt FRMD7 function through loss of a phosphorylation site and/or interference with protein-protein interactions. Both p.Q487X and p.V549Y fsX554 mutations were predicted to generate nonfunctional truncated proteins. Using a capture next generation sequencing method, we excluded CASK as the responsible gene for the remaining family. Combining sequence analysis and structural modeling, we report three novel mutations in FRMD7 in three independent families with XLICN and provide molecular insights for future XLICN diagnosis and treatment.
Similar content being viewed by others
Introduction
Idiopathic congenital nystagmus (ICN) causes involuntary and periodic ocular motility and often seriously reduces visual acuity. It is the most common oculomotor disorder characterized by bilateral uncontrollable ocular oscillation1, which usually presents in infancy or within the first few months after birth without any other sensory defect2. However, the inheritance pattern is heterogeneous, with autosomal dominant, autosomal recessive, or X-linked (XLICN) patterns, among which X-linked is the most common3,4. Genes causing XLICN have been mapped at three loci (Xp11.4–p11.3, Xp22 and Xq26–q27)1,5,6. XLICN, mapped to Xq26–q27, is linked to the FRMD7 gene7, which encodes a member of the protein 4.1 superfamily7. GPR143 resides at Xp22 and mutations within it are primarily linked to ocular albinism (OA), where nystagmus results as a secondary phenotype. However, GPR143 mutations have been reported in XLICN families, without the classical phenotype of ocular albinism8,9,10. Recently, Watkins et al. reported mutations in the CASK gene, locates at Xp11.4–p11.3, is linked with XLICN and mental retardation11. Taken together, so far, two genes (FRMD7 and GPR143) have been identified as causative genes for XLICN and CASK for XLICN with mental retardation, respectively.
Here, we recruited four families diagnosed with XLICN. Through direct sequencing for the two candidate genes, FRMD7 and GPR143, we identified three novel mutations in FRMD7 in three independent families. Meanwhile, we have excluded CASK, FRMD7 and GPR143 as the disease gene for the remaining family, which provides more evidence to show the genetic heterogeneity associated with XLICN and paves the way for discovering new disease genes for XLICN.
Results
Clinical data
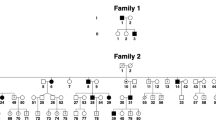
The patterns of inheritance of these four families are X-linked as indicated by the pedigrees (Figure 1). The clinical data are summarized in Table 1. Mental retardation, night blindness and photophobia are not observed in any of the affected individuals in any family, nor is there any incidence of systemic or other ocular anomalies.

Pedigrees of four families with X-linked congenital nystagmus.
Squares indicate males, circles indicate females and slashed symbols indicate deceased, black symbols indicate affected individuals, unfilled symbols indicate unaffected individuals and unaffected, obligate carriers are represented by a dotted circle. Arrow marks the proband.
Mutation identification and analysis
In family A (Figure 1), a novel missense mutation, c.780C > A, was identified at codon 780 (AGC to AGA) in exon 9 of FRMD7 gene. The c.780C > A mutation causes a substitution of serine to arginine at position 260 (p.S260R) of the FRMD7 protein (Figure 2). The p.S260R mutation occurred at a residue that is evolutionarily highly conserved (Figure 3). Results from online software prediction program strongly suggest that p.S260R variation is a pathogenic mutation. In family B, a nonsense mutation Q487X in exon 12 of FRMD7 introduces a premature stop codon and produces a truncated protein, which is missing the C-terminal 228 residues (Figure 2, 3). In family C, the deletion mutation (c.1645del G) in exon 12 of FRMD7, which is predicted to cause a frame shift at codon 549 and will stop the open reading frame at codon 554, thereby generating a prematurely truncated FRMD7 protein (Figure 2, 3). These mutations cosegregated with affected individuals or carriers in the family (A, B, C) and were not present in any of the unaffected family members (A, B, C) or 200 normal controls. The rest of the coding regions did not show any sequence changes.

Sequencing results of affected males and controls.
Three mutations of FRMD7 were identified in three families, which were c.780C > A (p.S260R) in family (A), c.1458 C > T (p.Q487X) in family (B), one G deletion mutation in family (C).

Multiple-sequence alignment of FRMD7 protein from different species.
The red outline in the alignment shows the amino acid affected by the mutation. The deletion mutation (bottom C), which leads to a frameshift mutation, the amino acid residues in red are the residues after frameshift mutation.
We searched these mutations in multiple databases, including the dbSNPs (v130), HapMap, 1000 Genome and 702 in-house exome data. The candidate mutations were not present in these databases. No mutation in FRMD7 was identified in family D and no mutations in GPR143 were detected in any individual among the four families.
We first manually checked of sequencing depth of CASK gene. On average, a mean coverage of 88X over the exons in this gene was achieved (Table S1). We further scanned the mutations in CASK gene and no mutations were identified (Table S1).
Taking this evidence together, we conclude that p.S260R, p.Q487X and p.V549YfsX554 are causative mutations in FRDM7, rather than benign polymorphisms in linkage disequilibrium with XLICN.
Discussion
ICN is the most common inherited eye disease manifesting with nystagmus and patients with this disease usually have problems with visual acuity. Here we recruited four families with XLICN, with the goal of identifying the genetic defects present in these families. Since only FRMD7 and GPR143 have been reported as causative genes for XLICN, we focused our attention on these two candidates. After comprehensive screening for mutations by direct sequencing, three novel mutations in FRMD7 were identified but no mutation was identified in GPR143. This study thus provides further evidence to show the role of FRMD7 in the pathogenesis of XLICN, which is consistent with previous evidence that 47% of the XLICN pedigrees in the Chinese population have yielded FRMD7 mutations12. Family D was also recruited but no FRMD7 or GPR143 mutations were identified in these individuals. Recently, there was a report that showed CASK may be one of the responsible genes for XLICN with mental retardation; we excluded it in the families of this study by capture next generation sequencing. These results further highlight the genetically heterogeneous nature of ICN, since at least four loci have been proposed for familial ICN. It also illustrates the unique role of FRMD7 in XLICN.
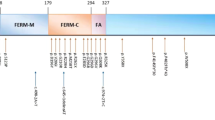
The FRMD7 protein (ENSP00000298542) consists of an N-terminal FERM domain, a FERM adjacent domain and a region without any significant homology to other proteins. The FERM domain is located between amino acids 2–282 and the FERM-C (F3) domain is located at residues 186–279 in FRMD7. Using the Phyre213 program, we predicted the secondary structure of the wild-type FRMD7 protein. We modeled the 3D structure of the wild type FRMD7 protein (1–336) and mapped out the S260 in the structure (Figure 4). The p.S260R mutation localizes to the FERM-C domain and introduces a larger amino acid (arginine) into a restricted area14. Since the S260 residue is solvent exposed, we rationalize that the point mutation (p.S260R) may potentially disrupt FRMD7 function through one of the following mechanisms. Firstly, S260 is predicted to be a phosphorylation site and p.S260R mutation will surely prevent the phosphorylation event and thus alter the regulation of FRMD715. Secondly, we mapped out the conserved regions of the FRMD7 surface. S260 is located in an extremely conserved surface that usually is involved in protein-protein interactions. It would not be surprising if any point mutation on this surface interferes with the interaction with other proteins and therefore contribute to the pathogenesis of the disease.

Structural model of the FRMD7 protein.
(A) The 3D model of FRMD7 (1–336) with the key mutation S260 colored in orange. (B) View of FRMD7 after 90 degree rotation. (C) The surface representation of B and the surface is color according to the conservation of the primary sequence. Green regions are strictly conserved, regions in purple are the least conserved areas and anything in-between is colored white. S260 locates on a large conserved surface.
The nonsense mutation and the deletion mutation in FRMD7 were predicted to cause gross defects at the protein level and loss of the protein function. To date, five mutations14,16,17,18 have been reported in exon 12 of FRMD7, all of which are predicted to produce truncated proteins. In the present study, two novel mutations in exon 12 were identified. Both are predicted to lead to premature termination of translation. It highlighted that the C-terminal region, which is a highly conserved domain, plays a critical role in the function of FRMD7 protein11.
The association between best corrected visual acuity (BCVA) and the type of XLICN mutations is of interest, since it may help the clinical genetic diagnosis. Since mutations in FRMD7 or GPR143 both can cause XLICN, we reviewed the literature to collect the available data of these two genes mutations and BCVA data, which was summarized in Table 2. We observed that patients with mutations in GPR143 have worse BCVA19,20,21, compared to those with mutations in FRMD72,22. Study data is consistent with the reported literature indicating a better BCVA in FRMD7 compared to GPR143 patients.
In summary, our findings have further expanded the mutational spectrum of FRMD7 and further confirmed the genetic heterogeneity of XLICN. By structural modeling, we predicted the consequence of the p.S260R mutation.
Meanwhile, from limited available data, it is suggested that XLICN patients with lower BCVA may have GPR143 gene mutations. However, the specific molecular consequence of these mutations in FRMD7 is not clear. Further studies are needed to provide insights into the detailed molecular pathogenesis of the mutations identified in the present study.
Methods
Clinical evaluations and DNA specimens
This study conformed to the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the Eye Hospital, Wenzhou Medical University. Written informed consent was obtained from all participating individuals or their guardians. Patients from four families (A, B, C and D, Figure 1) originating from the provinces of Helongjiang, Jilin, Inner Mongol and Shandong, respectively, were recruited. Clinical and ophthalmological examinations were performed by ophthalmologists.
Mutation analysis
Primers were designed to cover the sequences of all coding exons and splice junctions of FRMD7 and GPR143. The DNA sequences of FRMD7 and GPR143 were obtained from the GenBank database (NM_194277.1 and NM_000273.2). The primer sequences of FRMD7 have been published22 and the primers and amplification conditions for GPR143 are listed in Table 3.
PCR products were sequenced on an ABI PRISM 3730 DNA Sequencer (Applied Biosystems, Foster City, CA). Sequences were aligned and the mutations were detected using Mutation Surveyor software (Soft Genetics, USA, www.softgenetics.com). The effects of the mutations on the protein coding region (synonymous, missense, nonsense, frameshift, etc) were predicted by Exome-assistant22. The effects of the mutations on protein function were assessed by four online softwares, includeing Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), Pmut (http://mmb2.pcb.ub.es:8080/PMut/) and Panther (http://www.pantherdb.org/tools/csnpScoreForm.jsp).
A total of 7080 exons on the X chromosome (Table S2) were selected by a gene capture strategy using the GenCap custom enrichment kit (MyGenostics, Beijing), which has been previously described23. The enrichment libraries were sequenced on an Illumina Solexa HiSeq 2000 sequencer for paired read 100 bp. The analysis was described previously23. Briefly, using the Solexa QA the cutadapt (http://code.google.com/p/cutadapt/), SOAP aligner, BWA and GATK programs to retrieve and align to identify SNPs and insertions or deletions (InDels). SNPs and InDels were annotated using the exome-assistant program (http://122.228.158.106/exomeassistant). We selected the CASK gene (NM_001126054) for the mutation survey.
References
Cabot, A. et al. A gene for X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome Xp11.4–p11.3. Am J Hum Genet. 64, 1141–1146 (1999).
Hu, Y. et al. A novel splicing mutation of the FRMD7 gene in a Chinese family with X-linked congenital nystagmus. Mol Vis 18, 87–91 (2012).
Self, J. & Lotery, A. The molecular genetics of congenital idiopathic nystagmus. Semin Ophthalmol. 21, 87–90 (2006).
Thomas, S. et al. Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain. 131, 1259–1267 (2008).
Bassi, M. T. et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet. 10, 13–19 (1995).
Kerrison, J. B. et al. Congenital motor nystagmus linked to Xq26–q27. Am J Hum Genet. 64, 600–607 (1999).
Chishti, A. H. et al. The FERM domain: a unique module involved in the linkage of cytoplasmic proteins to the membrane. Trends Biochem Sci. 23, 281–282 (1998).
Watkins, R. J. et al. The Role of FRMD7 in Idiopathic Infantile Nystagmus. J Ophthalmol. 2012, 460956 (2012).
Zhou, P. et al. Identification of a novel GPR143 deletion in a Chinese family with X-linked congenital nystagmus. Mol Vis. 14, 1015–1019 (2008).
Liu, J. Y. et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet. 52, 565–570 (2007).
Watkins, R. J. et al. A novel interaction between FRMD7 and CASK: evidence for a causal role in idiopathic infantile nystagmus. Hum Mol Genet. 22, 2105–2118 (2013).
He, X. et al. A novel frameshift mutation in FRMD7 causing X-linked idiopathic congenital nystagmus. Genet Test. 12, 607–613 (2008).
Kelley, L. A. & Sternberg, M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 4, 363–371 (2009).
Tarpey, P. et al. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet. 38, 1242–1244 (2006).
Tsukita, S. & Yonemura, S. Cortical actin organization: lessons from ERM (ezrin/radixin/moesin) proteins. J Biol Chem. 274, 34507–34510 (1999).
Khan, A. O. et al. Prolonged Pursuit by Optokinetic Drum Testing in Asymptomatic Female Carriers of Novel FRMD7 Splice Mutation c. 1050 + 5 G > A. Arch Ophthal-Chic. 129, 936–940 (2011).
He, X. et al. A Novel Frameshift Mutation in FRMD7 Causing X-Linked Idiopathic Congenital Nystagmus. Genet Test. 12, 607–613 (2008).
Du, W. et al. A novel frame-shift mutation in FRMD7 causes X-linked idiopathic congenital nystagmus in a Chinese family. Mol Vis. 17, 2765–2768 (2011).
Hu, J. et al. A novel GPR143 splicing mutation in a Chinese family with X-linked congenital nystagmus. Mol Vis. 17, 715–722 (2011).
Yan, N. et al. A novel nonsense mutation of the GPR143 gene identified in a Chinese pedigree with ocular albinism. PLoS One. 7, e43177 (2012).
Preising, M. et al. Deletion in the OA1 gene in a family with congenital X linked nystagmus. Br J Ophthalmol. 85, 1098–1103 (2001).
Schorderet, D. F. et al. Novel mutations in FRMD7 in X-linked congenital nystagmus. Mutation in brief #963. Online. Hum Mutat. 28, 525 (2007).
Huang, X. F. et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One. 8, e63832 (2013).
Acknowledgements
We thank the families for their participation in this project. We are indebted to Dr. Jinyu Wu (Institute of Genomic Medicine, Wenzhou Medical University) for bioinformatics analysis. This work was supported by grants from the Chinese National Program on Key Basic Research Project (973 Program, 2013CB967502, F.G.), the Natural Science Foundation of China (81201181/H1818, F.G.), Zhejiang provincial & Ministry of Health research fund for medical sciences (WKJ2013-2-023, F.G.), Wenzhou Medical College PI starting grant (QTJ12011, F.G.).
Author information
Authors and Affiliations
Contributions
F.G. and J.Q. conceived the idea, X.Z., X.L.G., Y.Y., Y.L.Z., Y.M.W. and Y.L. collected the samples and performed the experiments, Z.B.J. and F.G. performed data analyses, X.Z. and J.S. performed structural modeling. F.G. wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhang, X., Ge, X., Yu, Y. et al. Identification of Three Novel Mutations in the FRMD7 Gene for X-linked Idiopathic Congenital Nystagmus. Sci Rep 4, 3745 (2014). https://doi.org/10.1038/srep03745
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03745
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
