
Abstract
Rhabdomyosarcoma (RMS) is the most commonly occurring type of soft tissue tumor in children. However, it is rare in adults and therefore, very little is known about the most appropriate treatment strategy for adult RMS patients. We performed genomic analysis of RMS cells derived from a 27-year-old male patient whose disease was refractory to treatment. A peritoneal seeding nodule from the primary tumor, pleural metastases, malignant pleural effusion and ascites obtained during disease progression, were analyzed. Whole exome sequencing revealed 23 candidate variants and 10 of 23 mutations were validated by Sanger sequencing. Three of 10 mutations were present in both primary and metastatic tumors and 3 mutations were detected only in metastatic specimens. Comparative genomic hybridization array analysis revealed prominent amplification in the 12q13–14 region and more specifically, the CDK4 proto-oncogene was highly amplified. ALK overexpression was observed at both protein and RNA levels. However, an ALK fusion assay using NanoString technology failed to show any ALK rearrangements. Little genetic heterogeneity was observed between primary and metastatic RMS cells. We propose that CDK4, located at 12q14, is a potential target for drug development for RMS treatment.
Similar content being viewed by others


Introduction
Rhabdomyosarcoma (RMS) is the most commonly occurring type of soft tissue tumor in children1, but it is less common in adults, accounting for only 2–5% of all soft tissue sarcomas2. Given the rarity of this disease, very little information is available on the most appropriate treatment strategy for adult RMS patients. Patients with unresectable or metastatic RMS have an extremely low cure rate and a poor prognosis3,4. A substantial improvement in survival has been achieved with the introduction of intensive chemotherapy regimens, which are usually based on pediatric oncology clinical trials on RMS5,6,7,8,9. However, survival rates for patients with metastatic disease remain disappointing and the prognosis is dismal in patients with a poor response to salvage chemotherapy3,10. Thus, identification of novel therapeutic targets in RMS is urgently needed in order to improve treatment outcomes for this aggressive type of tumor.
Major histologic subtypes of RMS include embryonal RMS (ERMS) and alveolar RMS (ARMS)11. Despite advances in therapy, patients with the ARMS histological variant of RMS have a 5-year survival of less than 30%. ARMS presents with distinctive chromosomal translocations that result in specific fusion gene products, the most prevalent of which are PAX3–FOXO1 (55%) and PAX7–FOXO1 (22%)12. Reciprocal translocation of chromosomes 2 and 13 results in a PAX3-FKHR fusion gene in ARMS, which fuses the region of the gene encoding the DNA-binding domain of the transcription factor PAX3 with that encoding the transactivation domain of the transcription factor FKHR in-frame (3, 4). However, at least 25% of ARMS cases lack such translocations, suggesting that ARMS is not a single disease, but a heterogeneous group of conditions with a common phenotype. Moreover, studies on the gene expression profile of RMS have proposed new molecular classifications13 and have revealed that a specific gene expression signature potentially determines tumor behavior as well as treatment outcome14,15,16. ALK is one of the targets of interest, given that ALK alterations are relatively common in RMS, although the function of its gene product remains unknown17.
Here, we report the clinical application of genomic profiling in identifying potential novel genetic mutations in patients with relapsed and chemotherapy-refractory alveolar RMS.
Results
Case presentation
A 27-year-old man presented with a complaint of left upper quadrant abdominal pain that had lasted for 3 years. Computed tomography (CT) and positron emission tomography (PET) scans showed multiple malignant masses, involving the pancreas and left upper abdominal wall and pleural seeding was also noted (Fig. 1A). Pathological examination of the abdominal wall mass showed thin fibrous septae lined by small round blue cells in an alveolar growth or solid pattern; the cells appeared to lack cohesion and had hyperchromatic nuclei and scant cytoplasm. The tumor cells were diffusely positive for CD99, desmin and WT1 and showed scattered focal positivity for cytokeratin. Ki-67 staining revealed high proliferative activity of the tumor cells. The FKHR break-apart fluorescence in situ hybridization (FISH) showed separate green and red signals, confirming FKHR rearrangement (Supplementary Fig. S1).

Computed tomography (CT) findings during the course of the disease.
(A). Abdominal wall mass (yellow) and pancreas mass (green) at the time of initial diagnosis. (B). After 1st-line chemotherapy, the disease virtually disappeared. (C). A massive amount of left pleural effusion was seen after salvage chemotherapy.
Based on the histology, immunohistochemistry (IHC) and FISH results, ARMS was diagnosed and alternating cycles of vincristine, doxorubicin and cyclophosphamide (VDC) and ifosfamide and etoposide (IE) were administered every 3 weeks.
After completing a 1-year course of cytotoxic chemotherapy, the patient achieved near complete remission, with disappearance of the multiple masses and pleural seeding (Fig. 1B). On the basis of a tumor board discussion involving a multi-modality team for sarcoma, the residual peritoneal seeding nodules were surgically resected. At the time of surgery, the resected seeding nodules were snap frozen and immediately stored at −80°C for molecular analysis. The pathologic examination of the resected peritoneal seeding nodules verified the diagnosis of ARMS.
Postoperative follow-up abdominal pelvis CT and chest CT demonstrated no evidence of malignancy. However, 3 months after surgical resection, the patient was found to have developed a chest wall mass of approximately 2 cm in size. Salvage etoposide, ifosfamide and cisplatin (VIP) chemotherapy was administered and initially elicited a partially positive response. However, soon afterward, the patient developed rapidly progressive disease with a massive amount of left pleural effusion after the 5th cycle of VIP (Fig. 1C). Because dyspnea was caused by rapidly increasing pleural effusion, talc pleurodesis and pleural biopsy were performed through video-assisted thoracoscopic surgery (VATS). At this time, the patient agreed to full genetic testing using the tissue specimens to identify potential molecular targets (sample #2).
In addition, malignant cells from the pleural effusion were cultured and stored at −80°C for molecular analysis and in vitro drug sensitivity tests (sample #3). However, the patient developed malignant ascites (sample #4) immediately after pleurodesis and 4 cycles of paclitaxel and ifosfamide were administered. The patient developed cord compression with paraplegia, experienced continued disease progression and died several weeks later.
Genomic profiling and somatic mutation
DNA from the primary tumor (sample #1) was sequenced, revealing 23 candidate variants: SCN1B, PPP1R3A, GRID2, APBA2, ZNF142, ZYG11A, RBFOX1, TCF7L1, NARF, KIAA0182, TEX13B, MUC2, LRRC3, GRHL3, MUC16, TTR, UBA1, FEN1, ELAC2, NBEAL1, DSCAML1, PCDHA4 and POLR3C (Table 1). Only the MUC16 mutation was detected in blood, with 3.4% allele frequency. Amino acid substitutions were predicted to arise from some of the point mutations in each gene and these in turn were predicted to have a substantial phenotypic effect based on the SIFT score (a SIFT score of 0 indicates a deleterious effect, a score ≤ 0.05 indicates a damaging effect and a score > 0.05 suggests that the substitution can be tolerated). These protein mutations were also predicted to have considerable functional impact based on the FI score as determined by Mutation Assessor (http://mutationassessor.org) (an FI score ≤ 0.8 is considered neutral; 0.8 < FI score ≤ 1.9 indicates low impact; 1.9 < FI score ≤ 3.5 indicates medium impact; and FI score > 3.5 indicates high impact). Among the variants detected in exome sequencing from the tumor specimen, those in SCN1B, PPP1R3A, GRID2, APBA2, ZNF142, ZYG11A, RBFOX1, TCF7L1, TEX13B and DSCAML1 were validated by Sanger sequencing and the details of these 10 candidate genes are provided in Table 2.
The primary tumor (sample #1), pleural metastases (sample #2) and malignant cells from ascites (sample #4) were all found to carry point mutations in SCN1B, PPP1R3A and ZYG11A. However, APBA2, ZNF142 and RBFOX1 mutations, although not present in the primary tumor, were detected in both metastatic (chemotherapy refractory) specimens. TCF7L1, TEX13B and DSCAML1 mutations, which were detected during exome sequencing, were not confirmed in subsequent Sanger sequencing of the primary tumor or any metastatic specimens. Although the mutation in GRID2 was not seen in the primary tumor due to failure of the sequencing reactions, the mutation was confirmed in both metastatic samples.
Comparative genomic hybridization array analysis of the primary tumor
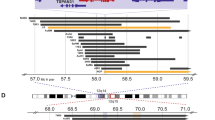
Although several chromosomal regions showed evidence of copy number variations (CNVs; Supplementary Table S1), the 12q13.3–q14.1 region demonstrated the highest level of chromosomal amplification (Fig. 2A). As this region contains multiple genes, we analyzed the amplification of each gene (Table 3). Within this region, the CDK4 proto-oncogene was highly amplified and several other genes in the amplicon also showed various degrees of amplification, including NACA, HSD17B6, SDR9C7, RDH16, GPR182, ZBTB39, TAC3, MYO1A, NAB2, STAT6 and LRP1. The overexpression of CDK4 at the protein level was also confirmed by IHC (Fig. 2B).

Array comparative genomic hybridization (aCGH) of the primary tumor and CDK4 overexpression.
(A). aCGH of the primary tumor: the 12q13.3–q14.1 region demonstrated the highest level of chromosomal amplification. (B-1). CDK4 immunohistochemistry of the primary tumor confirmed overexpression of CDK4 protein; CDK4 is located within the 12q13.3-q14.1 region. (B-2). Negative control for CDK4 immunohistochemistry.
ALK fusion assay
As ALK overexpression has been reported in refractory RMS18,19, we assessed the level of ALK protein expression using IHC. As shown in Fig. 3A-1, ALK IHC was strongly positive in most of the tumor cells. Given the high level of ALK protein expression, ALK RNA overexpression was also detected in the pleural metastasis specimen (sample #2), as expected (Fig. 3B).

ALK expression and fusion assay.
(A). ALK immunohistochemistry (2A-1, positive in the present case, 2A-2, negative control) and ALK FISH (2A-3) of the primary tumor: ALK protein overexpression was confirmed, but ALK rearrangement was not detected. (B). ALK RNA expression in tumor cells from pleural metastasis (sample#2): ALK RNA overexpression was detected. The ALK expression has been normalized to that of 4 housekeeping genes. Lung cancer cell lines (NCIH3122, NCIH2228 and A549) were used as controls for ALK RNA expression and EML-ALK fusion detection. (C). ALK fusion assay using NanoString: no EML4-ALK RNA was detected in the sarcoma specimen.
Two lung cancer cell lines, NCIH3122 and NCIH2228, were used as positive controls for EML4–ALK fusion and A549 cells were used as the negative control. ALK-fusion lung cancer only overexpresses the 3′-ALK mRNA (NCIH3122 and NCIH2228), whereas sarcoma overexpresses the full-length mRNA. The mean of 3′-ALK expression of ALK+ lung tumor is approximately 500, whereas this reached approximately 3000 in the sarcoma. Despite ALK overexpression at the protein and RNA levels, ALK amplification was not observed in the tumor cells of this patient.
Next, we screened for the presence of ALK fusion partners using a NanoString assay; however, the ALK fusion assay failed to show any ALK rearrangements (Fig. 3C). ALK rearrangement was also not detected by FISH (Fig. 3A-3).
Discussion
Patients with recurrent RMS usually present with a rapidly deteriorating condition and have markedly limited options in terms of chemotherapy9,20. In this study, we found that the majority of somatic mutations found during exome sequencing of primary tumor tissue were also observed in metastatic tumor tissue and metastatic cells in ascites samples, although Sanger sequencing revealed genetic alterations involving several genes, such as APBA2, ZNF1142 and RBFOX1, only in the metastatic samples. These results led to 2 important conclusions: First, there is little genetic heterogeneity between primary and metastatic RMS cells at least in terms of mutational spectra, reflecting relatively little genetic evolution during the course of metastasis. This is consistent with the results of recent similar studies on melanoma21, breast22 and pancreatic cancers23, in which genomic profiling for both primary and metastatic sites was performed. Second, we confirmed that malignant cells isolated from body fluid can be used for genomic profiling, as their genome is nearly identical to that of the resected tumor specimen. This may be especially important in clinical practice because body fluid can be obtained relatively easily using a bedside procedure. Of the 23 candidate genes found during exome sequencing, we selected 9 mutated genes (APBA2, RBFOX1, TCF7L1, MUC16, UBA1, FEN1, NBEAL1, DSCAML1 and PCDHA4) for which substantial functional impact was predicted (medium or high functional impact; FI score > 1.9), with or without damaging/deleterious phenotypic effects based on the SIFT score (≤0.05) and assessed their clinical relevance to RMS. However, we could not find any pre-existing evidence that these genetic alterations contribute to RMS development.
The aCGH array used in this study revealed prominent amplification in the 12q13 and 12q14 regions. Although we found that many genes within this chromosome 12 region were amplified, CDK4 amplification was of particular interest because it is known to play a pivotal role in the oncogenic process24 and perhaps more importantly, the corresponding proteins are potential drug targets25. Its overexpression is frequently observed in well-differentiated and dedifferentiated liposarcomas26,27,28; consequently, a clinical trial of the CDK4 inhibitor (PD0332991) for CDK4-amplified tumors has been conducted. In both phase I and phase II trials, the CDK4 inhibitor has proven effective in C and a randomized phase 3 trial is being considered by researchers29. Amplification of 12q13–q14 and CDK4 in RMS has been reported previously30,31, as has amplification of MYCN and both of these genes are known to be involved in RMS tumorigenesis32. However, these genes are associated with distinct expression profiles and clinical parameters. MYCN overexpression occurs more frequently in cases in which 2p24 amplification is present, whereas CDK4 overexpression is associated with 12q13–14 amplification33. In addition, 12q13–14 amplification was significantly associated with poor clinical outcomes, such as short failure-free and overall survival, compared to that seen in cases with 2q24 amplification33. In the RMS case studied here, we confirmed the presence of a 12q13–14 amplification and showed that CDK4 is one of the genes overexpressed in this chromosomal region.
A study provided in vitro evidence for the successful pharmacologic inhibition of CDK4/CDK6 activity in myoblasts and RMS-derived cells34; in this study, most ARMS-and ERMS-derived cell lines and tumor samples expressed CDK4 and CDK6 and exposure of these cells to a CDK4 inhibitor caused G1 cell cycle arrest, which is closely associated with myogenic differentiation. Given that defective cell cycle control, which leads to failure of myogenic differentiation, is one of the notable characteristics of RMS-derived cells, it was not surprising that CDK4 inhibition with PD0332991 ultimately facilitated skeletal muscle differentiation. This finding suggests that CDK4 inhibition is a potential therapeutic strategy for RMS. However, there is a scarcity of data on the use of a CDK4 inhibitor in patients with RMS. Although it is therefore difficult to draw firm conclusions regarding the potential efficacy of this inhibitor, the need for novel therapeutics arising from the dismal prognosis in refractory RMS, together with the genetic profiling data presented here, warrant clinical trials on a CDK4 inhibitor in chemotherapy-refractory RMS patients.
In agreement with previous reports18,19, we found that ALK was overexpressed in the RMS tumor in the current case. Although ALK overexpression is frequently detected in RMS, the mechanisms underlying this phenomenon are yet to be defined. However, a high-affinity binding site for the PAX3 and FOXO1 transcription factors in the intron of ALK has been reported to mediate high levels of ALK transcription35 and increases in ALK copy number have also been described17,18, although this did not always correlate with elevated ALK protein expression. A recent extensive cohort study on ALK aberration in RMS17 revealed that approximately 90% of ARMS patients and 50% of ERMS patients exhibited ALK copy number gains, whereas only 4% of RMS patients showed true amplification of ALK. In our study, ALK amplification was not observed, although ALK was overexpressed. The results of our study indicate that the overexpression of wild-type ALK alone may not be sufficient to drive tumor growth and that ALK may therefore not be an effective drug target in RMS. Currently, clinical trial NCT # 01121588 (clinicaltrials.gov) on crizotinib therapy for ALK-positive solid tumor types is ongoing. Since our study is limited to one case only, further studies are required to elucidate the antitumor efficacy of crizotinib and the CDK4 inhibitor in sarcomas in the context of clinical trials.
In summary, our study revealed that there was little genetic heterogeneity between primary and metastatic RMS cells and suggested that malignant cells from body fluid can be used for genomic profiling of RMS patients. The RMS tumor in this case overexpressed ALK, but this was not associated with the amplification or translocation of this gene. Prominent amplification of the 12q13–14 region was also observed and we propose that CDK4, located in 12q14, is a potential target for drugs in RMS.
Methods
Ethics statement
This study was approved by the SMC Institutional Review Board and was conducted in accordance with the 1996 Declaration of Helsinki. Written informed consent was obtained from the patient before genomic analyses were performed for research purposes.
IHC
Five-micrometer-thick tissue sections were deparaffinized in xylene, rehydrated and heated to 100°C in citrate buffer (pH 6.0) for 5 min for non-enzymatic antigen retrieval. The sections were incubated with monoclonal mouse anti-human desmin antibodies (1:100 dilution; RLM30; Novocastra, Newcastle-upon-Tyne, UK) for 60 min at room temperature, followed by incubation with a 1:1000 dilution of biotinylated goat anti-mouse IgG (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. The sections were stained with diaminobenzidine chromogen for 5–10 min and were then counterstained with hematoxylin for 5 min.
FISH
FISH was performed using commercially available ALK (Vysis LSI ALK Dual Color, Break Apart Rearrangement Probe; Abbott Molecular, Abbott Park, IL) and FKHR (Vysis LSI FKHR Dual Color, Break Apart Rearrangement Probe; Abbott Molecular) probes according to the manufacturer's instructions. One hundred cells were analyzed in each case. FISH was considered positive when more than 15% of the tumor cells showed distinct red and green signals and/or a single red (residual 3′) signal; alternatively, the specimen was classified as FISH negative.
Biospecimen processing and quality control
Excised tumor tissues were divided into 2 pieces. One piece was embedded in optimal cutting temperature compound and used to prepare hematoxylin and eosin-stained frozen section slides. The other pieces of tissue were snap frozen in liquid nitrogen and stored at −80°C. The tumor cell populations on the frozen section slide accounted for more than 60% of the total cell population; less than 10% were necrotic. Genomic DNA was extracted from snap frozen tissue and peripheral blood using the QIAmp DNA Mini kit (Qiagen GmbH, Hilden Germany) according to the manufacturer's instructions. DNA integrity was evaluated using 1% agarose gel electrophoresis. Tumor and normal DNA concentrations were measured using PicoGreen dsDNA Quantitation Reagent (Invitrogen, Carlsbad, CA). A minimum DNA concentration of 20 ng/μl was required for aCGH.
Exome sequencing and analysis
Genomic DNA was extracted from the blood and primary tumor (abdomen) of the RMS patient. Exon capture was performed using Agilent SureSelectXT Human All Exon (50 M), which includes all exons annotated in the consensus CDS (CCDS) database, as well as 10 bp of flanking sequence for each targeted region (http://www.genomics.agilent.com). The captured DNA fragments were sequenced with Illumine Hiseq2000, generating 100 bp × 2 paired-end reads. The clean reads were aligned against the human reference genome (hg19/GRCh37) using the Burrows-Wheeler Aligner (BWA). The alignment results were further processed sequentially using local realignment, duplicate read marking and base quality recalibration by using the Picard (http://picard.sourceforge.net) and GATK (http://www.broadinstitute.org/gatk/) pipeline software. Variant and germline calling were performed using JointSNVMix (http://code.google.com/p/joint-snv-mix/) and the somatic mutations observed in tumor tissue were annotated using ANNOVAR (http://www.openbioinformatics.org/annovar/).
Quality control, sequence alignment, somatic variant calling and annotation
In the first quality control step, Cutadapt v.1.0 [1] removed adapter sequences from the input fastq sequence. After adapter trimming, Fastx v.0.0.13 [2] filtered low-quality reads, such that base quality was more than 20 and the proportion of good-quality bases in each read was more than 50%. Finally, cmpFastq [3] classified paired-end reads and single-end reads. Classified fastq sequences were aligned to the human reference sequence (hg19) using the Burrow-Wheeler Aligner v.0.5.9 (BWA) [4] and were then merged to a BAM file. Subsequently, sequential cleanup processes, consisting of the addition or replacement of read groups, marking and removing duplicates and fixing mate information were performed using Picard Tools v.1.69 [5]. The cleaned bam file was then sorted using Samtools v.0.1.18 [6] and the local realignment and base quality score recalibration were processed using the Genome Analysis Toolkit v.1.6–7 (GATK) [7].
Somatic mutations were designated into 3 categories: single nucleotide polymorphisms (SNPs), indels and CNVs. We began by applying the joint_snv_mix_one model in JointSNVMix v.0.7.5 [8] in order to find point mutations and used Annovar [9], Mutation Assessor [10] and SIFT [11] for annotation. Annovar performed filter-based annotation indicating mutations that are present in 1000 genome projects or dbSNP (snp135). It also performed gene-based annotation using Mutation Assessor and SIFT to identify whether protein-coding changes caused by SNPs or CNVs are deleterious. We selected genes that were annotated as “medium or high functional impact” by Mutation Assessor and were predicted as “damaging” by SIFT. Indels were detected by the SomaticIndelDetector in GATK, following which Annovar gene-based annotation was used to describe the functional impact of somatic indels. CNVs were detected using ExomeCNV (R package) from the coverage file prepared using DepthOfCoverage in GATK. We used default parameters, except for the aforementioned software.
aCGH
Genomic DNA was extracted from the cells cultured from the primary tumor of the patient. aCGH was performed using the Agilent Human Genome CGH Microarray Kit 8 × 60 K, which contains approximately 45,000 probes.
ALK fusion transcript assay
nCounter assays were performed in duplicate, according to the manufacturer's instructions (NanoString Technologies, Inc, Seattle, WA, USA). Briefly, 500 ng of total RNA was hybridized to nCounter probe sets for 16 h at 65°C. Samples were processed using an automated nCounter Sample Prep Station (NanoString Technologies, Inc). Cartridges containing immobilized and aligned reporter complexes were subsequently imaged on the nCounter Digital Analyzer (NanoString Technologies, Inc), set at 1155 fields of view. Reporter counts were collected using the nSolver analysis software version 1 in NanoString, normalized and analyzed as described below. A detailed description of the assay is given elsewhere36.
References
Ognjanovic, S., Linabery, A. M., Charbonneau, B. & Ross, J. A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 115, 4218–4226 (2009).
Ferrari, A. et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 98, 571–580 (2003).
Esnaola, N. F. et al. Response to chemotherapy and predictors of survival in adult rhabdomyosarcoma. Ann. Surg. 234, 215–223 (2001).
Hawkins, W. G. et al. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer 91, 794–803 (2001).
Meza, J. L., Anderson, J., Pappo, A. S. & Meyer, W. H. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. J. Clin. Oncol. 24, 3844–3851 (2006).
McDowell, H. P. et al. Outcomes in paediatric metastatic rhabdomyosarcoma: results of The International Society of Paediatric Oncology (SIOP) study MMT-98. Eur. J. Cancer 46, 1588–1595 (2010).
Carli, M. et al. European intergroup studies (MMT4-89 and MMT4-91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J. Clin. Oncol. 22, 4787–4794 (2004).
Breneman, J. C. et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J. Clin. Oncol. 21, 78–84 (2003).
Pappo, A. S. et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J. Clin. Oncol. 17, 3487–3493 (1999).
Little, D. J. et al. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer 95, 377–388 (2002).
Parham, D. M. & Ellison, D. A. Rhabdomyosarcomas in adults and children: an update. Arch. Pathol. Lab. Med. 130, 1454–1465 (2006).
Sorensen, P. H. et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J. Clin. Oncol. 20, 2672–2679 (2002).
Davicioni, E. et al. Molecular classification of rhabdomyosarcoma--genotypic and phenotypic determinants of diagnosis: a report from the Children's Oncology Group. Am. J. Pathol. 174, 550–564 (2009).
Davicioni, E. et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 66, 6936–6946 (2006).
Reichek, J. L. et al. Genomic and clinical analysis of amplification of the 13q31 chromosomal region in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Clin. Cancer Res. 17, 1463–1473 (2011).
Missiaglia, E. et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 30, 1670–1677 (2012).
van Gaal, J. C. et al. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J. Clin. Oncol. 30, 308–315 (2012).
Corao, D. A. et al. ALK expression in rhabdomyosarcomas: correlation with histologic subtype and fusion status. Pediatr. Dev. Pathol. 12, 275–283 (2009).
Pillay, K., Govender, D. & Chetty, R. ALK protein expression in rhabdomyosarcomas. Histopathology 41, 461–467 (2002).
Mazzoleni, S. et al. Outcomes and prognostic factors after recurrence in children and adolescents with nonmetastatic rhabdomyosarcoma. Cancer 104, 183–190 (2005).
Turajlic, S. et al. Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Res. 22, 196–207 (2012).
Ding, L. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010).
Yachida, S. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010).
Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245 (1992).
Singer, S. et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 67, 6626–6636 (2007).
Louis-Brennetot, C. et al. The CDKN2A/CDKN2B/CDK4/CCND1 pathway is pivotal in well-differentiated and dedifferentiated liposarcoma oncogenesis: an analysis of 104 tumors. Genes Chromosomes Cancer 50, 896–907 (2011).
Wolf, M. et al. Microsatellite markers as tools for characterization of DNA amplifications evaluated by comparative genomic hybridization. Cancer Genet. Cytogenet. 93, 33–38 (1997).
Szymanska, J. et al. Gains and losses of DNA sequences in liposarcomas evaluated by comparative genomic hybridization. Genes Chromosomes Cancer 15, 89–94 (1996).
Dickson, M. A. et al. Phase II trial of the CDK4 inhibitor PD0332991 in CDK4-amplified liposarcoma. J. Clin. Oncol. 30 (Suppl), Abstr 10002 (2012).
Forus, A. et al. Mapping of amplification units in the q13-14 region of chromosome 12 in human sarcomas: some amplica do not include MDM2. Cell Growth Differ. 4, 1065–1070 (1993).
Anderson, J., Gordon, A., Pritchard-Jones, K. & Shipley, J. Genes, chromosomes and rhabdomyosarcoma. Genes Chromosomes Cancer 26, 275–285 (1999).
Missiaglia, E. et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer 48, 455–467 (2009).
Barr, F. G. et al. Genomic and clinical analyses of 2p24 and 12q13-q14 amplification in alveolar rhabdomyosarcoma: a report from the Children's Oncology Group. Genes Chromosomes Cancer 48, 661–672 (2009).
Saab, R. et al. Pharmacologic inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Mol. Cancer Ther. 5, 1299–1308 (2006).
Cao, L. et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 70, 6497–6508 (2010).
Lira, M. E. et al. Multiplexed Gene Expression and Fusion Transcript Analysis to Detect ALK Fusions in Lung Cancer. J. Mol. Diagn. 15, 51–61 (2013).
Acknowledgements
This work was supported by a grant from the Korea Healthcare Technology R&D project, Ministry for Health & Welfare Affairs, Republic of Korea (A092255).
Author information
Authors and Affiliations
Contributions
J.L. and Y.C. conceived the idea and designed the study. S.P. and J.L. collected and analyzed data and S.P. and I.D. wrote the main manuscript text. Y.C., I.D. and M.M. prepared Figures 2 and 3. J.J., K.R., S.A., L.M., S.K., K.K., M.M., J.K., E.O. and Y.K. contributed by providing study material. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Park, S., Lee, J., Do, IG. et al. Aberrant CDK4 Amplification in Refractory Rhabdomyosarcoma as Identified by Genomic Profiling. Sci Rep 4, 3623 (2014). https://doi.org/10.1038/srep03623
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03623
This article is cited by
-
Cyclins and cyclin-dependent kinases: from biology to tumorigenesis and therapeutic opportunities
Journal of Cancer Research and Clinical Oncology (2023)
-
Dll1+ quiescent tumor stem cells drive chemoresistance in breast cancer through NF-κB survival pathway
Nature Communications (2021)
-
Signaling pathways in Rhabdomyosarcoma invasion and metastasis
Cancer and Metastasis Reviews (2020)
-
Cyclin-dependent kinase 11p110 (CDK11p110) is crucial for human breast cancer cell proliferation and growth
Scientific Reports (2015)
-
The history and future of targeting cyclin-dependent kinases in cancer therapy
Nature Reviews Drug Discovery (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
