
Abstract
kefB is annotated as a potassium/proton antiporter in M. tuberculosis. There have been divergent reports on the involvement of KefB in phagosomal maturation in M. bovis BCG and no investigation has been carried out on its role in M. tuberculosis, the pathogenic species responsible for causing tuberculosis. This study was taken up to ascertain the involvement of KefB in the growth of M. tuberculosis and its role in phagosomal maturation and survival of the pathogen in guinea pigs. Our findings show that kefB mutant of M. tuberculosis (MtbΔkefB) was impaired i) for growth in high concentrations of potassium and ii) in arresting phagosomal acidification. However, the disruption of kefB had no adverse effect on the survival of M. tuberculosis in macrophages as well as in guinea pigs suggesting that the role of KefB in phagosomal acidification is unrelated to the survival of the pathogen in the host.
Similar content being viewed by others


Introduction
Phagocytosis of the invading microorganisms represents the first line of defense by macrophages triggering the signaling cascade of immune responses for subsequent killing of the ingested microbe1,2,3. An intricate mechanism follows the ingestion of microbe, which involves formation of the phagosomes4,5. These early phagosomes with pH ~ 6.5 are marked by the presence of Rab5 and early endosomal autoantigen 1 (EEA1) and are relatively poor in proteases, however, their fusion with various endosomal compartments leads to the formation of late phagosomes marked by the presence of Rab7, Rab9, lysobisphosphatidic acid, mannose-6-phosphate receptor and a low pH of ~5.54,5. Consequently, the fusion of late phagosomes and lysosomes [which is marked by the presence of lysosome-associated membrane proteins (LAMPs)] triggers the release of several hydrolytic enzymes responsible for killing the pathogen4,5. However, intracellular pathogens such as Mycobacterium tuberculosis are endowed with various strategies that help them in subverting the phagosomal maturation pathways and survive in the macrophages in spite of the host assault6. These strategies prevent acidification of M. tuberculosis containing phagosomes beyond pH ~ 6.5, restrict phagolysosomal fusion and provide the pathogen with an environment that is conducive for its survival in the host6,7,8. Several components of M. tuberculosis have been demonstrated for their involvement in arresting phagosomal maturation9,10,11,12,13,14,15,20. Lipid products of M. tuberculosis such as lipoarabinomannan (LAM) and phosphatidylinositol mannoside (PIM) are necessary to accomplish inhibition of phagolysosomal fusion9,10,11. This involves binding of mannose-capped LAM to mannose receptors on macrophages for the incorporation of the former into macrophage cell membranes and thereby affecting the host signaling platform resulting in the altered function of protein kinases and cytokine production that is necessary for vesicle fusion and phagosomal maturation11. Additionally, glycosylated LAM and PIM by their intercalation into the host membranes have been demonstrated to inhibit the acquisition of EEA-1, syntaxin 6 and cathepsin D, the molecules which are required for the normal membrane fusion involved in the phagosome trafficking9,10,11. M. tuberculosis PtpA, a secreted protein tyrosine phosphatase contributes towards the inhibition of phagosome-lysosome fusion by dephosphorylating and inactivating the host vacuolar protein sorting 33B (VPS33B) (a member of the class C VPS complex that regulates membrane fusion within the endocytic pathway)12. PtpA also binds to subunit H of macrophage V-ATPase and blocks its activity required for phagosome acidification13. Another M. tuberculosis protein namely Nucleoside Diphosphate kinase (Ndk) exhibits GAP (GTPase activating protein) activity against Rab5 and Rab7 leading to their inactivation by depleting the γ-phosphate from GTP bound Rab proteins required for phagosomal maturation14. Another M. tuberculosis protein SecA2 (an export system protein) has been proposed for its involvement in the phagosomal maturation based on the increased acidification of phagosomes containing secA2 mutant of M. tuberculosis in comparison to the phagosomes containing the parental strain15. M. tuberculosis SapM, a secretory phosphatase, has also been shown to be essential for phagosomal maturation by virtue of its ability to dephosphorylate PI3P that is required by the phagosomes for docking of Rab effector proteins important for phagolysosomal fusion16,17,18,19,20.
kefB has been annotated as potassium/proton antiporter in M. tuberculosis. However, there has been no study to ascertain whether KefB has any role in the growth of M. tuberculosis as a function of potassium concentration. In a transposon mutagenesis based strategy to identify phagosomal acidification defective mutants of M. bovis BCG, kefB was found to prevent acidification of phagosomes as kefB mutants of M. bovis BCG were observed to accumulate in the acidified phagosomes21. However, in another study, by employing a targeted kefB mutant of M. bovis BCG, it was demonstrated that the kefB mutant could arrest the phagosomal maturation similar to the parental M. bovis BCG strain22. Moreover, this kefB mutant displayed enhanced survival in comparison to the parental M. bovis BCG when grown in host macrophages22. Thus, there have been divergent observations about the role of KefB in the phagosomal maturation arrest in M. bovis BCG. More importantly, in M. tuberculosis, the pathogenic species responsible for causing tuberculosis, the role of KefB has not been investigated. Hence, in this study, we aimed to ascertain the role of KefB in the growth, phagosomal maturation and pathogenesis of M. tuberculosis by employing a kefB mutant of the pathogen.
Results
Construction and characterization of kefB mutant of M. tuberculosis
To evaluate the role of KefB in the physiology and pathogenesis of M. tuberculosis, we constructed a kefB mutant of M. tuberculosis (MtbΔkefB) as described in the methods. Disruption of kefB was confirmed by three approaches (Fig. 1A). Firstly, the kefB gene was amplified from M. tuberculosis and MtbΔkefB genomic DNA by employing the primers KefB-F-NdeI and KefB-R-NdeI. While a product of 1.1 kb corresponding to the size of kefB was obtained in the case of M. tuberculosis, a band of 2.2 kb was amplified in the case of MtbΔkefB confirming the disruption of the gene due to insertion of the hygromycin resistance cassette (Fig. 1B). Secondly, we employed another set of primers which confirmed the homologous recombination of the allelic exchange substrate (AES) at the legitimate position in the genome of M. tuberculosis. For this, we employed primers KefB-up and Hyg-up which yielded a product of 961 bp and primers KefB-Dn and Hyg-Dn which yielded a product of 909 bp (Fig. 1C and 1D). Thirdly and more importantly, the amplification products were subjected to DNA sequencing that further confirmed the site specific recombination as well as disruption of kefB in MtbΔkefB. A complemented strain MtbΔkefBComp was generated by the introduction of plasmid pVR1-prokefB into the electro-competent cells of MtbΔkefB. For confirming the genetic complementation of kefB gene, pVR1-prokefB plasmid was isolated from MtbΔkefBComp cells followed by the digestion of the plasmid by NdeI restriction enzyme. The resulting 1.1 kb band observed on agarose gel during electrophoresis confirmed the presence of kefB gene in MtbΔkefBComp (Fig. 1E).

Characterization of MtbΔkefB mutant.
(A) Representation of the genomic arrangement of the disrupted kefB gene and the depiction of the primer pairs employed for the characterization. (B) Confirmation of kefB gene deletion in M. tuberculosis by PCR by using the gene specific primers KefB-F-NdeI and KefB-R-NdeI (marked by red arrows in Fig. 1A) to obtain 1.1 kb amplification in M. tuberculosis (lane 3) and 2.2 kb amplification in MtbΔkefB (lane 4). 100 bp ladder and λHindIII were loaded in lanes 1 and 2, respectively. (C) Confirmation of kefB gene deletion in M. tuberculosis by PCR by using the primers KefB-Dn and Hyg-Dn (marked by green arrows in Fig. 1A). Lane 1 shows a 909 bp amplification in MtbΔkefB, lane 2 – 100 bp ladder (D) Confirmation of kefB gene deletion in M. tuberculosis by PCR by using the primers KefB-up and Hyg-up (marked by blue arrows in Fig. 1A). Lane 1 – 100 bp ladder and lane 2 shows a 961 bp amplification in MtbΔkefB. (E) Confirmation of complementation of the M. tuberculosis kefB mutant. The presence of pVR1-prokefB was confirmed by restriction digestion of the plasmid isolated from the complemented strain to yield a 1.1 kb band of kefB gene along with its promoter (lane 3). λHindIII and 100 bp ladder were loaded in lanes 1 and 2, respectively.
kefB mutant of M. tuberculosis shows significant growth defect in high concentrations of potassium
KefB is annotated as a potassium efflux pump which supposedly acts as a potassium/proton antiporter effluxing out potassium ions and taking in hydrogen ions in exchange. Hence, the loss of KefB pump is expected to render M. tuberculosis sensitive to the levels of potassium which we assessed by growing M. tuberculosis, MtbΔkefB and MtbΔkefBComp in the presence of varying concentrations of potassium and monitoring the growth of mycobacteria. As M. tuberculosis faces ~ 30–50 mM concentration of potassium in the phagosomes23,24,25, the growth kinetics was compared at 0 mM, 7 mM, 50 mM and 125 mM concentrations of potassium. In the case of complete absence of potassium, all three strains apparently exhibited a bit poor growth although in comparison to each other, there was no difference in the growth rate of any of the three strains (Fig. 2A). At 7 mM concentration of potassium, the growth of all three strains showed improvement when compared with the growth at 0 mM potassium concentration; however, the disruption of kefB did not appear to make any difference as the growth rate of all the three strains was comparable (Fig. 2B). These observations show that the disruption of kefB has no effect on the growth of M. tuberculosis at low potassium concentrations. In contrast, however, the role of KefB in the growth of M. tuberculosis became apparent when the strains were grown in high potassium concentrations. At 50 mM and 125 mM concentrations of potassium, the MtbΔkefB grew similar to the parental strain until ~ mid-logarithmic phase after which the mutant exhibited an impaired growth as it transitioned to the stationary phase at a much lower A600 nm when compared with M. tuberculosis or MtbΔkefBComp (Fig. 2C and 2D).

Growth kinetics of M. tuberculosis, MtbΔkefB and MtbΔkefBComp in broth culture in the presence of varying concentrations of potassium.
Media were inoculated with M. tuberculosis, MtbΔkefB or MtbΔkefBComp with a starting absorbance (A600 nm) of 0.02 and the growth was monitored daily in the presence of 0 mM (A), 7 mM (B), 50 mM (C) or 125 mM (D) concentration of potassium. The values of absorbance are represented as the mean (±SE) of three independent experiments. The data was analyzed by two-way analysis of variance (ANOVA) with the Bonferroni multiple comparison test (***p < 0.001).
Disruption of kefB reduces the ability of M. tuberculosis to arrest phagosomal maturation
As no studies have been carried out on the involvement of KefB in arresting phagosomal maturation in M. tuberculosis, we employed FITC labeled M. tuberculosis, MtbΔkefB and MtbΔkefBComp and studied the localization of the pathogen in the acidic compartments by using LysoTracker Red dye. The latex beads, used as control for the experiment, underwent ~87% colocalization with the LysoTracker rich compartments. M. tuberculosis exhibited only 22.8% colocalization with the LysoTracker labeled acidified compartments, however, the mutant strain with disruption of kefB exhibited a substantially higher (61.5%) colocalization with the acidified compartments (Fig. 3). MtbΔkefBComp, the complemented strain, exhibited 16% colocalization with the LysoTracker labeled acidified compartments which was not significantly different from the corresponding value observed in the case of M. tuberculosis (Fig. 3). These observations demonstrate the role of KefB in the phagosomal maturation. Additionally, we analyzed the colocalization of FITC labeled M. tuberculosis, MtbΔkefB and MtbΔkefBComp with the early endosomes by employing antibodies against Rab5, which is an early endosomal marker. It was observed that M. tuberculosis and MtbΔkefBComp accumulated in the Rab5 containing early endosomes resulting in an effective arrest of phagosome-lysosome fusion while MtbΔkefB and latex beads exhibited very poor colocalization with Rab5 indicating their inability to arrest the phagosome maturation (Fig. S1).

Influence of kefB disruption on the ability of M. tuberculosis to arrest phagosomal acidification.
(A) RAW264.7 macrophages were infected with FITC labeled M. tuberculosis, MtbΔkefB, MtbΔkefBComp and latex beads (green) separately. The cells were then subjected to LysoTracker Red staining followed by observation under confocal microscope. Representative fluorescent images depict that disruption of kefB leads to the accumulation of MtbΔkefB in acidified organelles (overlap of green and red images appears yellow as shown by arrows) while the M. tuberculosis and MtbΔkefBComp were found in non-acidified organelles. Latex beads employed as a positive control display colocalization with the LysoTracker rich compartments. The scale bars depict 5 μm. (B) Bar diagram represents the percentage of phagosomes containing M. tuberculosis, MtbΔkefB, MtbΔkefBComp and latex beads that colocalized with LysoTracker Red. Data is the mean (±SE) of 3 independent experiments carried out in triplicates (***p < 0.001, One- way ANOVA).
kefB mutant of M. tuberculosis exhibits no adverse effect on its ability to survive in macrophages
To assess the ability of M. tuberculosis, MtbΔkefB and MtbΔkefBComp to survive in macrophages, RAW 264.7 macrophages were separately infected with all three strains with an MOI of 5:1 and the growth was monitored at various time points. We observed that M. tuberculosis and MtbΔkefBComp grew normally inside the macrophages until 6 days post-infection in a comparable manner. MtbΔkefB also exhibited similar growth pattern initially for 2 days post-infection, however, thereafter the mutant strain displayed an enhanced growth when compared with the parental strain and at the end of 6 days post-infection, the intracellular CFU of MtbΔkefB was ~2 fold higher when compared with M. tuberculosis and MtbΔkefBComp (Fig. 4).

Intracellular growth of M. tuberculosis, MtbΔkefB and MtbΔkefBComp in RAW 264.7 macrophages.
RAW 264.7 cells were infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp at an MOI of 5:1 (bacteria:macrophages). The number of intracellular viable bacteria was determined at various time points. A significant difference in the growth of MtbΔkefB in comparison to M. tuberculosis was observed on the 6th day of infection. The values are represented as the mean (±SE) of three independent experiments carried out in triplicates. The data was analyzed by two-way analysis of variance (ANOVA) with the Bonferroni multiple comparison test (***p < 0.001).
Disruption of kefB exhibits no effect on the growth of M. tuberculosis in guinea pig model of infection
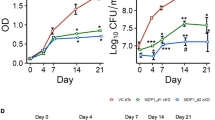
The importance of kefB in the growth and pathogenesis of M. tuberculosis in the host was determined by employing guinea pig model of experimental tuberculosis. Guinea pigs were infected with 10–30 bacilli of M. tuberculosis, MtbΔkefB or MtbΔkefBComp by using the aerosol route of infection and euthanized at 5 weeks (Fig. 5A) and 10 weeks (Fig. 5B) post-infection. We observed no significant difference in the pulmonary bacillary load of all the three strains at 5 weeks post-infection which corresponded to 5.03 log10CFU, 5.04 log10CFU and 5.07 log10CFU in the case of infection with M. tuberculosis, MtbΔkefB or MtbΔkefBComp, respectively. Similarly, the splenic bacillary load amongst the guinea pigs infected with these strains was also comparable with 4.84 log10CFU, 4.88 log10CFU and 4.80 log10CFU in the case of infection with M. tuberculosis, MtbΔkefB or MtbΔkefBComp, respectively (Fig. 5A). On extending the time period to 10 weeks also, the bacillary load in the organs of the animals infected with M. tuberculosis or MtbΔkefB or MtbΔkefBComp was comparable. The bacillary load in the lungs of the guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp was 5.13 log10CFU, 4.97 log10CFU and 5.06 log10CFU, respectively, while the bacillary load in the spleens was found to be 5.16 log10CFU, 5.43 log10CFU and 5.02 log10CFU, respectively (Fig. 5B). These observations show that KefB does not play a vital role in the pathogenesis of M. tuberculosis in guinea pig model of experimental tuberculosis.

Influence of disruption of kefB on the growth of M. tuberculosis in guinea pigs.
The figure depicts bacillary load in the lungs and spleen of guinea pigs infected with 10–30 bacilli of M. tuberculosis, MtbΔkefB or MtbΔkefBComp at 5 weeks (A) and 10 weeks post-infection (B). Guinea pigs infected with either of the M. tuberculosis strains exhibited a comparable bacillary load in the lungs as well as in the spleen. Each data point represents the log10CFU value for an individual animal and bar depicts mean (±SE) for each group.
Guinea pigs infected with M. tuberculosis or MtbΔkefB exhibit comparable pathology and survival time
The organs of the guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp were analyzed for gross pathological as well as for histopathological differences. At 5 weeks post-infection, the gross pathological changes in the organs of guinea pigs infected with all the three strains were comparable. The lungs of these animals displayed moderate involvement with occasional large tubercles whereas liver and spleen displayed numerous small sized tubercles (Fig. 6A). This was further substantiated by the histopathological analyses (Fig. 6B). The lungs of the infected animals from all groups exhibited comparable granulomatous inflammation encompassing a large proportion of the total lung area with a severe loss of the lung parenchyma while the liver tissues displayed effacement of a large proportion of the hepatic parenchyma along with granulomatous infiltration. At 10 weeks post-infection, the lungs, liver and spleen of the guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp exhibited extensive pathological changes which were more severe than those observed at 5 weeks. However, comparable pathology was observed in animals infected with all three strains with no significant differences in the gross pathological scores between the organs (Fig. 7A). Both lungs and spleens of the infected animals displayed inflammation and heavy involvement marked by the presence of numerous large tubercles (Fig. 7A). Scattered areas of necrosis were also observed both in the liver and spleen of the infected animals. Moreover, the extent of histopathological changes was much more pronounced than that observed at 5 weeks post-infection, however, a comparative analysis amongst the animals from different groups revealed that the pathological changes in the organs of the animals infected with various M. tuberculosis strains were comparable as no significant differences could be discerned (Fig. 7B). The lung sections exhibited severe lung pathology characterized by multiple coalescing granulomas and widespread lymphocytic infiltration resulting in complete loss of pulmonary micro-architecture. Similarly, the liver sections also displayed extensive pathological changes with large areas covered by multi-focal granulomas (Fig. 7B). Together, our findings on bacillary load and pathological changes demonstrate that the disruption of kefB did not influence the pathogenesis of M. tuberculosis in guinea pigs.

Gross pathological and histopathological changes in the organs of infected guinea pigs at 5 weeks post-infection.
(A) The figure depicts representative photographs of gross pathological lesions and graphical depiction of gross scores of lungs, liver and spleen of guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp and euthanized at 5 weeks post-infection. The organs of guinea pigs infected with all the strains exhibited comparable pathology. No significant differences were observed in the gross pathological scores for the lungs, liver and spleen of guinea pigs infected with any of the strains. Each data point represents the score of an individual animal and the bars depict medians (±interquartile ranges) for each group. (B) The figure depicts representative 40× photomicrographs of hematoxylin and eosin (H&E) stained 5 μm sections of lung and liver of guinea pigs euthanized at 5 weeks post-infection. The histopathological changes observed in the lung and liver of guinea pigs infected with MtbΔkefB were similar when compared with the changes observed in M. tuberculosis or MtbΔkefBComp infected animals exhibiting numerous foci of granulomatous infiltration. The scale bars depict 200 μm.

Gross pathological and histopathological changes in the organs of infected guinea pigs at 10 weeks post-infection.
(A) The figure depicts representative photographs of gross pathological lesions and graphical depiction of gross scores of lungs, liver and spleen of guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp and euthanized at 10 weeks post-infection. Heavy involvement and extensive pathology was observed in the organs of guinea pigs infected with any of the strains. No significant differences were observed in the gross pathological scores for lungs, liver and spleen of guinea pigs infected with any of the strains. Each data point represents the score of an individual animal and the bars depict medians (±interquartile ranges) for each group. (B) The figure depicts representative 40× photomicrographs of hematoxylin and eosin (H&E) stained 5 μm sections of lung and liver of guinea pigs euthanized at 10 weeks post-infection. No difference was observed in the histopathological changes in the lung and liver of guinea pigs infected with MtbΔkefB when compared with the histopathological changes observed in M. tuberculosis or MtbΔkefBComp infected animals. The scale bars depict 200 μm.
When the survival of guinea pigs infected with M. tuberculosis, MtbΔkefB or MtbΔkefBComp strains was monitored, we found that there was no difference in the median survival time of the animals infected with any of the three strains (Fig. 8). We observed that the median survival time of M. tuberculosis infected guinea pigs was 130.5 days (Fig. 8). The guinea pigs infected with MtbΔkefB also exhibited a comparable median survival time of 144 days (Fig. 8). A median survival time of 117.5 days as observed in the case of guinea pigs infected with MtbΔkefBComp confirmed that the disruption of kefB does not influence the disease causing ability and pathogenesis of M. tuberculosis in guinea pigs (Fig. 8).

Influence of disruption of kefB gene of M. tuberculosis on the survival of guinea pigs post-infection.
Guinea pigs (8 per group) were aerogenically infected with 10–30 bacilli of either M. tuberculosis, MtbΔkefB or MtbΔkefBComp and monitored for survival post-infection. The median survival time (MST) for animals in each infected group is stated in brackets.
Discussion
M. tuberculosis is a formidable pathogen which successfully exploits the host systems for its own survival. Arresting the phagosomal acidification and maturation is one of the important features of M. tuberculosis that turns phagosomes into a niche for the survival and replication of the pathogen7,8,26,27,28. Multiple mycobacterial factors have been reported for their involvement in subverting phagolysosomal fusion in M. tuberculosis9,10,11,12,13,14,15,20. Due to divergent observations about the involvement of kefB in arresting phagosomal maturation in M. bovis BCG and lack of studies on its function in the pathogenic M. tuberculosis, we aimed to ascertain the influence of KefB on the growth of M. tuberculosis, if any and evaluate its role in arresting phagosomal maturation by employing a kefB mutant of the pathogen.
First, to evaluate the influence of KefB on the growth of M. tuberculosis, we compared the ability of M. tuberculosis with MtbΔkefB as well as the complemented strain MtbΔkefBComp to survive in broth culture with various potassium concentrations ranging from 0 to 125 mM. While no difference in the growth pattern amongst the three strains was observed at 0 and 7 mM potassium, at higher potassium concentrations (50 mM and 125 mM) the mutant exhibited impaired growth by transiting to stationary phase at much lower A600 nm when compared with the parental or the complemented strain, which supports the proposed role of KefB as a potassium efflux pump. At higher potassium concentrations, the presence and function of potassium efflux pump would be necessary to reduce the increasing intracellular potassium concentration resulting from the influx of potassium in order for it to prevent the cell from potassium toxicity. Thus, for the first time, we show that KefB is important for the growth of M. tuberculosis at high potassium concentrations.
Further, the role of kefB in the phagosomal maturation arrest was studied by employing M. tuberculosis along with the kefB mutant and the complemented strain in colocalization experiments by using LysoTracker Red. When macrophages were infected with M. tuberculosis, as expected, the bacilli exhibited very less colocalization with the LysoTracker Red dye thus confirming that the pathogen significantly inhibited the phagosomal acidification and resided majorly in non-acidified phagosomes with only ~22% of the bacilli in the acidified phagosomes. However, the mutant strain was unable to prevent the phagosomal acidification to the same magnitude as the parental strain as was evident from the observation that 62% of the mutant bacteria resided in the acidified compartments confirming thereby the involvement of KefB in preventing phagosomal acidification. The MtbΔkefBComp strain exhibited similar results as observed in the case of M. tuberculosis with only ~16% of the bacilli residing in acidified phagosomes. The most likely mechanism for KefB to mediate phagosomal acidification arrest could stem from the antiporter nature of this pump wherein potassium can be effluxed out into the phagosomal lumen and protons can be taken inside the bacterial cytoplasm for preventing the acidification of phagosomal lumen, hence, in the absence of KefB, acidification of phagosomes can continue. However, contrary to our expectations MtbΔkefB exhibited better survival in macrophages when compared with the parental and the complemented strains. These observations suggest that bacterial death is not necessarily the only outcome resulting from the acidification of phagosomes. Infact, several examples do exist wherein the acidification of phagosomes does not influence the growth of the pathogen in macrophages21,29,30. Transposon mutants of M. tuberculosis Rv2693c and mmpL9 did not show any attenuation of growth in macrophages in spite of their trafficking to late endosomal compartments29. In another study, M. tuberculosis mutants that were incapable of phagosomal acidification arrest were identified and two of these mutants (in the genes Rv1522c and Rv2930) displayed normal intracellular growth and behaved similar to the wild type M. tuberculosis30. In another study by Stewart et al., it was observed that a few BCG mutants exhibited normal survival and growth in macrophages in spite of losing their ability to inhibit phagosomal maturation21. Hence, the acidification of phagosomes does not necessarily seal the fate of the pathogen; it could escape death in spite of acidification which could possibly mean that the mutation in such cases although related to acidification of phagosomes does not necessarily influence the survival of the pathogen.
Further, when M. tuberculosis, MtbΔkefB or MtbΔkefBComp were evaluated for their growth and pathogenesis in guinea pigs, we observed that guinea pigs infected with all three strains separately exhibited comparable bacillary load in the lungs and spleens and also there was no difference in the pathological changes. The guinea pig model of low-dose, air-borne tuberculosis infection with virulent M. tuberculosis is the most widely employed model for the elucidation of events in the pulmonary tuberculosis pathogenesis. Infection of guinea pigs with a low dose of <10 CFU of virulent M. tuberculosis results in the dissemination of the pathogen from lungs to pulmonary lymph nodes within 10 to 12 days post-infection via hematogenous spread and appears in spleen ~3 weeks post-infection, after which secondary pulmonary granulomas are formed by reseeding of the bacilli in the lungs by ~4 weeks post-infection31,32. The comparable bacillary load in the organs of the guinea pigs infected with either M. tuberculosis or MtbΔkefB indicates that KefB may not be indispensable for the growth and pathogenesis of M. tuberculosis in guinea pigs. Our studies in RAW 264.7 macrophages also support these observations.
In conclusion, for the first time, we show that M. tuberculosis KefB, the proposed potassium efflux pump, is important for maintaining the normal growth pattern of M. tuberculosis in high potassium concentrations. We also demonstrate the involvement of KefB in preventing the acidification of phagosomes. However, the lack of kefB in M. tuberculosis has no adverse effect on its ability to survive in macrophages and guinea pigs suggesting that the role of KefB in the acidification of phagosomes is unrelated to its survival in host.
Methods
Bacterial strains and growth conditions
Escherichia coli strains XL-1 Blue and HB101 were used for cloning and were grown in Luria Bertani (LB) broth or on LB agar. Mycobacterial strains were grown in Middlebrook (MB) 7H9 broth supplemented with 10% ADC, 0.2% glycerol and 0.2% tween 80 with constant shaking at 200 rpm or on MB7H11 agar supplemented with 10% OADC and 0.2% glycerol at 37°C. Kanamycin and chloramphenicol were used at a concentration of 25 μg/ml and 30 μg/ml, respectively. Hygromycin was employed at a concentration of 50 μg/ml for mycobacteria or at 150 μg/ml for E.coli.
Disruption of kefB (Rv3236c) and genetic complementation of the mutant
Primers KefB-AI-F (5′gatatcggtacccgccgcgaggtgtcgatgttg3′) and KefB-AI-R (5′gaattctctagagaatcggcgacaacgcgaatc3′) were designed to amplify Amplicon I (695 bp), comprising of 100 bp 5′ proximal region of kefB and 595 bp sequence immediately upstream to kefB, while the primers KefB-AII-F (5′gaattcctcgaggccacggcgtatgtgttcgtc3′) and KefB-AII-R (5′gatatcactagtcgatcgatgatgatgctggaggg3′) were employed for the amplification of Amplicon II (691 bp), comprising of 96 bp of 3′ distal region of kefB and 595 bp sequence immediately downstream to kefB. The amplicons I and II were PCR amplified and cloned into the vector pYUB854 flanking the hygromycin resistance cassette at KpnI/XbaI and XhoI/SpeI, respectively, to generate pYUBΔkefB. A 3.4 kb fragment (ΔkefB::hyg), was excised from pYUBΔkefB by employing KpnI/SpeI and the resulting linear Allelic Exchange Substrate (AES) was electroporated into M. tuberculosis as described earlier20,33 to generate the kefB mutant of M. tuberculosis (MtbΔkefB). For the confirmation of kefB disruption, KefB-F-NdeI (5′ggattacatatggtggaggtttcgagggcg3′), KefB-R-NdeI (5′ggattacatatgctagttcgaagctactgcggc3′), KefB-Dn (5′ttagcccgaccacttcac3′), Hyg-Dn (5′aaatcagatatcggacaagc3′), KefB-up (5′gatcatgctcggattcgtc3′) and Hyg-up (5′cgcacacataaaaacagtgc3′) primers were employed.
For the genetic complementation of MtbΔkefB, the kefB gene and its promoter were cloned into pVR134 as follows. A 500 bp region comprising the promoter region of kefB gene was PCR amplified by using the primers Pro-KefB-F-XbaI (5′ggattatctagagatgacggcggggcacttgctt3′) and Pro-KefB-R-SphI (5′ggattagcatgccgcacccagaatctgagc3′) and then cloned into the vector pVR1 at the promoter cloning sites XbaI/SphI resulting in the plasmid pVR1-pro. The kefB gene was then PCR amplified by using the primers KefB-F-NdeI and KefB-R-NdeI and the resulting amplicon was cloned into pVR1-pro at NdeI restriction site downstream to the cloned promoter. The resulting plasmid pVR1-prokefB was subjected to sequencing and was introduced into MtbΔkefB mutant, after confirming the sequence of the promoter region as well as kefB gene, by electroporation to generate the complemented strain MtbΔkefBComp.
Growth kinetics under various conditions
M. tuberculosis, MtbΔkefB and MtbΔkefBComp strains were grown in minimal media (MM) with defined potassium concentrations and the growth was monitored daily by measuring the absorbance at 600 nm. MM was prepared by adding 0.5% asparagine (wt/vol), 2% glycerol, 0.5 mg of ZnCl2 liter−1, 0.1 mg of MnSO4 liter−1 and 40 mg of MgSO4 liter−1 in double distilled water and desired concentrations of KH2PO4.
Infection of RAW 264.7 macrophages and bacterial enumeration
M. tuberculosis, MtbΔkefB and MtbΔkefBComp strains were grown in MB7H9 media containing 0.2% tween 80, 10% ADC and appropriate antibiotics. Exponentially growing cells were harvested at ~4,000 × g for 10 minutes and washed twice with MB7H9 media. Resulting bacterial cells were resuspended in 10 ml of RPMI media containing 10% FCS to which ~6 gram of 0.5 mm glass beads were added followed by vortexing for 15 minutes. The suspension was centrifuged at 50 × g in order to remove any remaining bacterial clumps. The CFU of the resulting homogeneous single cell suspension of bacteria was estimated by measuring the absorbance of this suspension at 600 nm (A600 nm of 0.5 corresponds to 3 × 107 M. tuberculosis CFU/ml under our culturing conditions).
RAW 264.7 cells were grown in RPMI media containing 10% FCS and antibiotic-antimycotic mix. The macrophage cells were scrapped by using a sterile scrapper and were counted by using trypan blue. This suspension of cells was infected with M. tuberculosis at an MOI of 5:1 (bacteria:macrophages) in RPMI media containing 10% FCS at 37°C for 2 hours with a constant shaking at 100 rpm. The cells were then harvested by centrifugation to ensure the removal of any non-phagocytosed extracellular bacteria and washed once with RPMI media. The remaining extracellular bacilli, if any, were killed by the addition of 200 μg/ml amikacin at 37°C for 1 hour with a constant shaking at 100 rpm. Finally, two rounds of washing with RPMI media were carried out to get rid of the remaining extracellular bacteria. 105 infected cells were then seeded in each of the 96 well microtiter plates in a final volume of 250 μl. The plates were then kept at 37°C in the presence of 5% CO2. On day 0, 2, 5 and 6, the infected macrophage monolayers were lysed with 200 μl of 0.025% SDS to release intracellular mycobacteria, which were then enumerated by plating appropriate dilutions on MB7H11 agar. Colonies were counted after 4 weeks of incubation at 37°C and CFU/ml was calculated.
Study of phagosomal maturation by using confocal microscopy
M. tuberculosis strains were labeled with Fluorescein isothiocyanate (FITC) (Sigma, MO, USA). Briefly, the M. tuberculosis cultures were grown to A600 nm of 0.5. The culture was harvested, washed twice with 0.5 M sodium bicarbonate buffer (pH 9.5) and resuspended in the same buffer supplemented with 100 μg/ml FITC followed by an overnight incubation at 4°C. Thereafter, the bacteria were pelleted, washed twice with PBS (pH 7.4) and single cell suspension was made as described above. Commercially available fluorescent latex beads (Diameter - 1.0 μm, Sigma, MO, USA.) were used separately as control. Macrophages were seeded on poly-L-lysine (Sigma, MO, USA) coated glass coverslips within a 12-well plate at a density of 5 × 105 macrophages per well and infected separately with 2.5 × 106 FITC labeled mycobacteria (ratio of 5:1, bacteria:macrophages). After a 4 h incubation, the cells were washed twice with fresh RPMI media and treated with 200 μg/ml amikacin for 2 h at 37°C to remove extracellular bacteria. Subsequently, the cells were incubated with 50 nM LysoTracker Red DND-99 (Invitrogen Life Technologies, CA, USA) in RPMI (supplemented with 10% FBS) for 1 h. After this, the cells were washed once with fresh RPMI media (supplemented with 10% FBS) and fixed with 4% paraformaldehyde in PBS. Coverslips were mounted by using ProLong Gold antifade reagent (Invitrogen Life Technologies, CA, USA) and analyzed by using Leica TCS SP5 confocal laser scanning microscope (Leica Microsystems, Mannheim, Germany). Subsequently, the fraction of FITC labeled mycobacteria that colocalized with LysoTracker Red was determined by analyzing ~100 phagosomes20.
For Rab5 staining, the protocol employed was similar as described above until amikacin treatment. Subsequently, cells were fixed with 4% paraformaldehyde in PBS for 16 hours. Blocking was carried out with 2% FBS for 1 hour at room temperature on a rocker followed by incubation with 5 μg/ml Anti-Rab5 antibodies (Biovision Research Prodcuts, CA, USA, raised in rabbit) for 3 hours at room temperature on a rocker. Cells were washed twice with PBS and further incubated with Texas Red-AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., PA, USA) at 1:1500 dilution for 1 hour at room temperature on a rocker. Cells were washed with PBS and the coverslips were mounted as described above.
In vivo guinea pig experiments
Pathogen-free out-bred female guinea pigs of the Duncan-Hartley strain in the weight range of 250–350 grams were obtained from the Disease Free Small Animal House Facility, Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar, India. To study the influence of kefB disruption on the growth and pathogenesis of M. tuberculosis, guinea pigs were infected by the aerosol route with 10 to 30 bacilli of either M. tuberculosis, MtbΔkefB or MtbΔkefBComp. Animals (n = 7) were euthanized at 5 weeks and 10 weeks post-infection by CO2 asphyxiation. After dissecting the animals, lungs, liver and spleen were scored for gross pathological changes such as the extent of involvement of the organs, number and size of the tubercles, areas of inflammation and changes due to necrosis. The gross pathological scores were graded from 1–4 based on the modified Mitchison scoring system described earlier35. Left caudal lung lobe and caudal segment of spleen from the infected animals were aseptically removed for bacterial enumeration. The specific segments of lung and spleen were weighed and homogenized separately in 4 ml saline by using a polytron homogenizer. Appropriate dilutions of the homogenates were plated on MB7H11 agar plates in duplicates and incubated at 37°C for 3–4 weeks. Colonies were counted and expressed as mean log10 CFU/organ. For histopathological evaluation, the right lung and a portion of left dorsal lobe of liver from the infected animals were removed and fixed in 10% buffered formalin. 5 μm thick sections from the formalin fixed, paraffin embedded tissues were stained with haematoxylin and eosin (H&E). The tissues were coded and the coded samples were analyzed by a certified pathologist having no knowledge of the experimental groups.
To study the influence of disruption of kefB on the survival of infected animals, guinea pigs (n = 8) were infected as described above with either M. tuberculosis, MtbΔkefB or MtbΔkefBComp and the median survival time of animals was measured.
Ethics statement
Protocols for all the animal experiments included in this manuscript along with the requirement of guinea pigs were reviewed and approved by the Institutional Animal Ethics Committee of University of Delhi South Campus, New Delhi, India (Ref. No. 1/IAEC/AKT/BIOCHEM/UDSC/14.10.2011). All animals were routinely cared for according to the guidelines of the CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals). The guinea pigs were euthanized by CO2 asphyxiation and all efforts were made to minimize animal suffering.
Statistical analysis
For comparing the growth of M. tuberculosis strains under i) various in vitro conditions and ii) in RAW 264.7 macrophages, two-way analysis of variance (ANOVA) with the Bonferroni multiple comparison test was employed. For the phagosomal maturation in RAW 264.7 macrophages and comparison of bacillary load in the lungs or spleen of infected guinea pigs, one-way ANOVA with the Tukey post test was employed. For comparison of the gross pathological scores of various groups, the nonparametric Kruskal-Wallis test was employed. Differences were considered significant when P < 0.05. For the statistical analysis and generation of graphs, Prism 5 software (version 5.01; GraphPad Software Inc., CA) was used.
References
Goldstein, E., Lippert, W. & Warshauer, D. Pulmonary alveolar macrophage. Defender against bacterial infection of the lung. J Clin Invest. 54, 519–528 (1974).
Greenberg, S. & Grinstein, S. Phagocytosis and innate immunity. Curr Opin Immunol. 14, 136–145 (2002).
Peterson, P. K., Verhoef, J., Schmeling, D. & Quie, P. G. Kinetics of Phagocytosis and Bacterial Killing by Human Polymorphonuclear Leukocytes and Monocytes. J Infect Dis. 136, 502–509 (1977).
Bouvier, G., Benoliel, A. M., Foa, C. & Bongrand, P. Relationship between phagosome acidification, phagosome-lysosome fusion and mechanism of particle ingestion. J Leukoc Biol. 55, 729–734 (1994).
Via, L. E. et al. Arrest of Mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem. 272, 13326–13331 (1997).
Ehrt, S. & Schnappinger, D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 11, 1170–1178 (2009).
Deretic, V. & Fratti, R. A. Mycobacterium tuberculosis phagosome. Mol Microbiol. 31, 1603–1609 (1999).
Deretic, V., Via, L. E., Fratti, R. A. & Deretic, D. Mycobacterial phagosome maturation, rab proteins and intracellular trafficking. Electrophoresis. 18, 2542–2547 (1997).
Fratti, R. A., Chua, J., Vergne, I. & Deretic, V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A. 100, 5437–5442 (2003).
Chua, J., Vergne, I., Master, S. & Deretic, V. A tale of two lipids: Mycobacterium phagosome maturation arrest. Curr Opin Microbiol. 7, 71–77 (2004).
Welin, A. et al. Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect Immun. 76, 2882–2887 (2008).
Bach, H., Papavinasasundaram, K. G., Wong, D., Hmama, Z. & Av-Gay, Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe. 3, 316–322 (2008).
Wong, D., Bach, H., Sun, J., Hmama, Z. & Av-Gay, Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A. 108, 19371–19376 (2011).
Sun, J. et al. Mycobacterial nucleoside diphosphate kinase blocks phagosome maturation in murine RAW 264.7 macrophages. PLoS One. 5, e8769 (2010).
Sullivan, J. T., Young, E. F., McCann, J. R. & Braunstein, M. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect Immun. 80, 996–1006 (2012).
Fratti, R. A., Backer, J. M., Gruenberg, J., Corvera, S. & Deretic, V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 154, 631–644 (2001).
Purdy, G. E., Owens, R. M., Bennett, L., Russell, D. G. & Butcher, B. A. Kinetics of phosphatidylinositol-3-phosphate acquisition differ between IgG bead-containing phagosomes and Mycobacterium tuberculosis-containing phagosomes. Cell Microbiol. 7, 1627–1634 (2005).
Saleh, M. T. & Belisle, J. T. Secretion of an acid phosphatase (SapM) by Mycobacterium tuberculosis that is similar to eukaryotic acid phosphatases. J Bacteriol. 182, 6850–6853 (2000).
Vergne, I. et al. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 102, 4033–4038 (2005).
Puri, R. V., Reddy, P. V. & Tyagi, A. K. Secreted Acid Phosphatase (SapM) of Mycobacterium tuberculosis Is Indispensable for Arresting Phagosomal Maturation and Growth of the Pathogen in Guinea Pig Tissues. PLoS One. 8, e70514 (2013).
Stewart, G. R., Patel, J., Robertson, B. D., Rae, A. & Young, D. B. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 1, 269–278 (2005).
Butler, R. E., Cihlarova, V. & Stewart, G. R. Effective generation of reactive oxygen species in the Mycobacterial phagosome requires K+ efflux from the bacterium. Cell Microbiol. 12, 1186–1193 (2010).
Wagner, D. et al. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis- and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J Immunol 174, 1491–1500 (2005).
Wagner, D. et al. Changes of the phagosomal elemental concentrations by Mycobacterium tuberculosis Mramp. Microbiology 151, 323–332 (2005).
Wagner, D. et al. Elemental analysis of the Mycobacterium aviumphagosome in Balb/c mouse macrophages. Biochem Biophys Res Commun 344, 1346–1351 (2006).
Vergne, I., Chua, J., Singh, S. B. & Deretic, V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol. 20, 367–394 (2004).
Deretic, V. et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 8, 719–727 (2006).
Deretic, V. et al. Phosphoinositides in phagolysosome and autophagosome biogenesis. Biochem Soc Symp. 74, 141–148 (2007).
MacGurn, J. A. & Cox, J. S. A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect Immun. 75, 2668–2678 (2007).
Pethe, K. et al. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci U S A. 101, 13642–13647 (2004).
McMurray, D. N. Disease model: pulmonary tuberculosis. Trends Mol. Med. 7,135–137 (2001).
McMurray, D. N. Guinea pig model of tuberculosis. In: Bloom B. R. (ed.), Tuberculosis: pathogenesis, protection and control. ASM Press, Washington, DC. 135–147 (1994).
Van Kessel, J. C. & Hatfull, G. F. Recombineering in Mycobacterium tuberculosis. Nat Methods 4, 147–152 (2007).
Reddy, P. V. et al. Disruption of Mycobactin Biosynthesis Leads to Attenuation of Mycobacterium tuberculosis for Growth and Virulence. J Infect Dis. 10.1093/infdis/jit250 (2013).
Jain, R. et al. Enhanced and enduring protection against tuberculosis by recombinant BCG-Ag85C and its association with modulation of cytokine profile in lung. PLoS One. 3, e3869 (2008).
Acknowledgements
We thank Praveen Kumar for help with the animal experiments. Rupangi Verma Puri and Priyanka Chauhan are acknowledged for critical reading of the manuscript. We thank Priti Singh and Tannupriya Gosain for excellent technical help. We thank Dr. Ashok Mukherjee for providing his services for histopathological analysis of the samples. This work was supported by a research grant from the Department of Biotechnology, Government of India.
Author information
Authors and Affiliations
Contributions
G.K. and A.K.T. conceived and designed the experiments. G.K., V.R. and P.S. performed the experiments. G.K. analyzed the data. G.K. and A.K.T. wrote the manuscript. A.K.T. provided overall supervision throughout the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Khare, G., Reddy, P., Sidhwani, P. et al. KefB inhibits phagosomal acidification but its role is unrelated to M. tuberculosis survival in host. Sci Rep 3, 3527 (2013). https://doi.org/10.1038/srep03527
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03527
This article is cited by
-
Mycobacterium tuberculosis sensor kinase DosS modulates the autophagosome in a DosR-independent manner
Communications Biology (2019)
-
A novel Tn1696-like composite transposon (Tn6404) harboring bla IMP-4 in a Klebsiella pneumoniae isolate carrying a rare ESBL gene bla SFO-1
Scientific Reports (2017)
-
Ciliate Paramecium is a natural reservoir of Legionella pneumophila
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
