
Abstract
The proinflammatory cytokine Interleukin 17A (hereafter named IL–17A) or IL-17A producing cells are elevated in breast tumors environment and correlate with poor prognosis. Increased IL-17A is associated with ER(−) or triple negative tumors and reduced Disease Free Survival. However, the pathophysiological role of IL-17A in breast cancer remains unclear although several studies suggested its involvement in cancer cell dissemination. Here we demonstrated that a subset of breast tumors is infiltrated with IL-17A-producing cells. Increased IL-17A seems mainly associated to ER(−) and triple negative/basal-like tumors. Isolation of tumor infiltrating T lymphocytes (TILs) from breast cancer biopsies revealed that these cells secreted significant amounts of IL-17A. We further established that recombinant IL-17A recruits the MAPK pathway by upregulating phosphorylated ERK1/2 in human breast cancer cell lines thereby promoting proliferation and resistance to conventional chemotherapeutic agents such as docetaxel. We also confirmed here that recombinant IL-17A stimulates migration and invasion of breast cancer cells as previously reported. Importantly, TILs also induced tumor cell proliferation, chemoresistance and migration and treatment with IL-17A-neutralizing antibodies abrogated these effects. Altogether these results demonstrated the pathophysiological role of IL-17A-producing cell infiltrate in a subset of breast cancers. Therefore, IL-17A appears as potential therapeutic target for breast cancer.
Similar content being viewed by others


Introduction
Inflammation often occurs in the microenvironment of tumors and actively takes part to the tumor progression process by favoring tumor cell survival and growth, angiogenesis and metastasis1. Interleukin 17A (hereafter named IL-17A) is a pro-inflammatory cytokine that belongs to a family encompassing 6 interleukins (IL-17A to F)2. IL-17A binds to a receptor composed of IL-17RA and IL-17RC dimer whose expressions are ubiquitous. IL-17A is mainly produced by a subset of CD4+ lymphocytes called Th17 cells. However, other cell types were reported to produce IL-17A including macrophages, dendritic cells, γδ T cells, NK and NKT cells, CD8+ T cells and neutrophils3,4. In humans, increased IL-17A is associated with infections, chronic inflammatory diseases and autoimmunity3. IL-17A or IL-17A-producing cells are also increased in malignancies5 including breast cancers6,7,8,9,10. In fact, the tumors cells and tumor-associated fibroblasts secrete factors and generate a pro-inflammatory cytokine milieu that leads to the recruitment of Th17 cells in the tumor microenvironment8. IL-17A producing cells thereby represent a subpopulation within the TILs from breast cancer8 and infiltration with IL-17A-producing immune cells is a poor prognosis factor10. A recent study indicated that infiltration with IL-17A+ immune cells is mainly observed in estrogen receptor negative (ER(−)), progesterone receptor negative (PR(−)) and triple negative tumors and associated with high histological grade and reduced disease free survival (DFS)10. It is therefore important to elucidate the pathophysiological role of IL-17A in breast cancer. It was previously shown that IL-17A may favor breast tumor cell dissemination6 and may be required for the growth of a murine breast tumor cell line in vivo11. Yet, the pro-oncogenic effect of IL-17A in breast cancer has not been thoroughly investigated and we thus decided to elucidate the functional role of IL-17A and IL-17A producing TILs in human breast cancers.
Results
IL-17A producing cells infiltrate human breast cancer biopsies
We first assessed expression of IL-17A by IHC in 40 breast cancer cases, 10 metastases and 10 matched controls. Whereas no or few IL-17A+ cells were observed in the normal tissues, 8 out of 40 (20%) of the cancer cases showed moderate to strong infiltration with IL-17A+ immune cells, mostly lymphocytes and macrophages, in the tumoral stroma (Figure 1, Supplementary Figure 1 and Table 1). There was a trend of association between the presence of IL-17A+ lymphocytes and tumors with ER(−) status (88% of IL-17A+ tumors were ER(−)). Conversely, the majority (92%) of ER(+) tumors were not infiltrated with IL-17A producing cells. Yet, the number of cases was too small to reach statistical significance. Among the 8 tumors that were infiltrated with IL-17A+ cells, 7 were ER(−), including 4 triple negative breast cancers and 1 Her2+ tumor. These observations are in line with the recent study from Chen and colleagues10 who reported that 18% of the 207 breast cancer cases analyzed were infiltrated with IL-17A producing cells. The presence of IL-17A was significantly associated with ER(−)/PR(−) tumors (P < 0.01) and triple negative (P < 0.05) tumors.

Representative Immunohistochemical staining of IL-17A expression in normal and breast cancer human tissues.
IL-17A stained sections of 40 invasive ductal breast carcinomas, 10 metastases and 10 matched normal counterparts. Brown staining indicates IL-17A protein. Arrows indicate IL-17A positive cells within the stroma, # refers to the corresponding sample in Supplementary Table 1.
In order to further demonstrate that IL-17A is released by lymphocytes infiltrating ER(−) breast cancers, we isolated and expanded tumor-infiltrating lymphocytes (TILs) from 6 ER(−) breast cancer biopsies. Biopsies were obtained following surgical procedures of breast cancer patients. 4 patients had a triple negative tumor and 2 patients had a Her2+ tumor. Tumor biopsies were collected and preserved in culture medium for subsequent isolation and separation of the different cell populations. The T lymphocytes were then expanded ex vivo as described in materials and methods section. Results revealed a phenotypic heterogeneous T lymphocyte population isolated from these biopsies. As illustrated in Figure 2, we could obtain significant IL-17A-secreting TILs in 4 out of the 6 TILs. Patient AL is a 29 year-old patient who presented with a triple negative, basal-like, pT2N0, SBR3 grade tumor. When isolated, the TILs from this patient were CD3+ lymphocytes, mostly (75%) CD4+ and secreted large amounts of IL-17A. Patient CP is a 40 year-old woman with a triple negative, basal-like, pT3N3a, SBR3 grade tumor. The tumor was infiltrated with a mixed population of CD3+ TILs that were CD4+, CD8+ or CD4+CD8+ and secreted IL-17A. Patient 432 is a 78 year-old woman with a relapsing triple negative, basal-like, pT4bNx, SBR3 grade breast cancer. The biopsy was infiltrated with TILs that secreted moderate amounts of IL-17A and were CD3+ (100%) and mostly CD8+ (90%) T cells. Patient 452 is a 52 year-old woman with and ER(−), PR(−) and Her2+, pT4bN1 and SBR3 grade breast cancer. The TIL population was mostly CD3+ (96%), CD4+ (70%) and secreted IL-17A. The expanded TILs of the 2 other patients, PR, a 66 year-old patient with a triple negative, apocrine, SBR3 grade, pT2N0 breast cancer and MAR, a 42 year-old woman with an ER(−), PR(−) and Her2+, SBR3 grade, pT3N1 tumor, did not secrete IL-17A ex vivo. It should be noted that in all cases, we could expand from the tumor biopsies between 1 to 3% of TCRγδ expressing lymphocytes. In aggregates, IHC and clinical data along with previous work published by others demonstrate that breast cancers are infiltrated with IL-17A producing immune cells. Such infiltration is particularly frequent in ER(−) tumors and more specifically in triple negative/basal-like tumors.

Characterization of lymphocyte subpopulations isolated from ER(−) breast cancer biopsies.
(A) Tumor infiltrating lymphocytes (TILs) were isolated from biopsies then short-term amplified ex vivo. Expression of CD3, CD4, CD8 and TCRγδ receptors were analyzed by flow cytometry. (B) Production of IL-17A by TILs was measured by ELISA in cell culture supernatants of 16 h cultures. Quantification was performed in duplicates.
IL-17A activates the ERK1/2 pathway in breast cancer cells
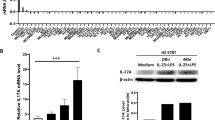
To address the functional role of IL-17A, we stimulated various human breast cancer cell lines with recombinant human IL-17A. Of note, all the human breast cancer cell lines tested expressed IL17RA and IL17RC (Supplementary Figure 2) the two monomers that form the functional receptor of IL-17A2. Therefore, all the breast cancer cell lines are able to respond to IL-17A stimulation. Putative signaling pathways activated by IL-17A were identified using anti-phospho tyrosine whole blot analysis. We identified several putative protein kinases recruited by IL-17A in the MCF7 and T47D breast cancer cell lines that could correspond to FAK, p70 S6 kinase, c-Raf and ERK1/2 according to their molecular weight (data not shown). However, when validated with specific antibodies, only ERK1/2 was consistently shown to be recruited by recombinant IL-17A in all the cell lines tested as illustrated in MCF7, T47D, MDA-MB468, MDA-MB157 and BT20 cells (Figure 3).

IL-17A activates ERK1/2 pathway in breast cancer cells.
Western blot analysis of phospho (pThr202/pTyr204) ERK1/2 and total ERK1/2 in several breast cancer cell lines untreated (medium) or treated with 10 ng/ml of recombinant IL-17A for 20 min (IL-17A). Data are representative of two independent experiments for each cell line.
IL-17A promotes resistance to docetaxel via activation of ERK1/2 pathway
We demonstrated here that IL-17A recruited the ERK1/2 pathway in breast cancer cells. As ERK kinases are involved in resistance to taxane-based therapies12, we tested whether IL-17A would mediate resistance to conventional chemotherapeutic agents such as docetaxel. To this end, human breast cancer cell lines were stimulated with recombinant human IL-17A and subsequently incubated with the drug. As illustrated in Figure 4, IL–17A decreased docetaxel-induced cell death in MCF7, T47D, BT20, MDA-MB468 and MDA-MB157 breast cancer cells in a dose-dependent manner. To delineate the role of ERK1/2 in IL-17A-mediated resistance to chemotherapeutic agents, we treated the cells with the MEK Inhibitor U0126. U0126 is a chemically synthesized organic compound that blocks the phosphorylation of ERK1/2 by inhibiting the kinase activity of MAP Kinase Kinase (MAPKK or MEK 1/2)13. U0126 inhibited IL-17A-induced phosphorylation of ERK1/2 (Figure 5B) and abrogated IL-17A-mediated protection from docetaxel-induced cell death in MCF7 (Figure 5A) whereas MEK inhibitor alone did not affect cytotoxicity (data not shown). U0126 also inhibited phosphorylation of ERK1/2 (Figure 5D) and abrogated resistance to docetaxel in BT20 cells stimulated with IL-17A (Figure 5C). Altogether these data demonstrate that IL–17A protects from docetaxel-induced cell death through activation of the ERK1/2 kinases and may therefore participate to therapy-resistance in breast cancer.

IL-17A promotes resistance to docetaxel.
MCF7, T47D, BT20, MDA-MB468 and MDA-MB157 cells were cultured for 48 h in complete medium alone (medium) or treated with recombinant human IL–17A at 1 or 10 ng/ml as indicated. Cells were then culture in FCS free medium supplemented with corresponding concentration of cytokine for 24 h and then further supplemented with docetaxel at 5, 10, 20 or 40 μg/ml as indicated for 7 h. The cytotoxicity was determined using the Cytotoxicity Detection Kit (Roche). For each cell line, data are representative of 3 independent experiments performed in duplicates (* P < 0.05; ** P < 0.01; *** P < 0.001).

IL-17A-induced resistance to docetaxel is dependent of ERK1/2 activation.
(A and C) MCF7 and BT20 cells were stimulated as described in Figure 4. When indicated, the MEK inhibitor U0126 was added at 10 μM 24 h before docetaxel. The cytotoxicity was determined using the Cytotoxicity Detection Kit (Roche). For each cell line, data are representative of 3 independent experiments performed in duplicates (* P < 0.05; ** P < 0.01). (B and D) Western blot analysis of phospho (pThr202/pTyr204) ERK1/2 and total ERK1/2 in MCF7 and BT20 cell lines untreated (medium) or treated with 10 μM U0126 inhibitor alone (U0126), 10 ng/ml of recombinant IL-17A (IL-17A) or 10 ng/ml of recombinant IL-17A + 10 μM U0126 inhibitor (IL-17A + U0126) for 30 min and 3 h.
IL-17A increases the proliferation of some breast cancer cell lines
IL-17A activated the ERK kinases in breast cancer cells and it was reported to stimulate the proliferation of human airway smooth muscle cells via this pathway14. We therefore investigated whether IL-17A could also enhance the proliferation of breast cancer cell lines. The proliferation of the T47D cells requires ERK recruitment (see Supplementary Figure 6 from ref15) and IL-17A indeed stimulated their proliferation in a dose-dependent manner (Figure 6). In contrast, the MCF7 cell line, which was found to be less sensitive to ERK-recruitment for prolactin-inducing proliferation15, failed to respond to IL-17A. IL-17A did not enhance the proliferation of BT20 and MDA-MB468 cells. In conclusion, IL-17A can increase the proliferation of some breast cancer cells potentially through the ERK1/2 pathway.

IL-17A increases proliferation of T47D cells.
MCF7, T47D, BT20 and MDA-MB468 breast cancer cell lines were cultured in complete medium supplemented with 0, 1, 10 or 100 ng/ml of recombinant human IL-17A as indicated. Cell proliferation was assessed at 72 h using tritiated thymidine ([3H]-TdR) incorporation protocol. Data are the mean ± SEM of two independent experiments, each performed in hexaplicates (* P < 0.05).
IL-17A promotes breast cancer cell migration and invasion
In our attempt to unravel the oncogenic properties of IL-17A in breast cancer, we tested whether IL-17A would promote migration and invasion as reported by others. Previous work indeed demonstrated that IL-17A stimulates migration and invasion in several cancer types16,17,18 including breast cancer6, thereby favoring metastasis. As shown in Figure 7A, IL-17A increased the migration of the poorly motile and non-invasive MCF7 cell line in a dose-dependent manner. Although IL-17A also increased the invasive ability of MCF7 cells in Boyden chambers assays (data not shown), the majority of IL-17A-treated-MCF7 cells remained non-invasive. Therefore, to evaluate the ability of IL-17A to foster cell invasion of the surrounding extracellular matrix, we used the MDA-MB231 cell line which endows intrinsic invasion capacities. As shown in Figure 7B, IL-17A increased invasion of MDA-MB231 cells in Boyden chambers assays. Increased invasive ability of IL-17A-treated MDA-MB231 cells was then further demonstrated using 3D Clusters assays (Figure 7C). As expected, MDA-MB231 cells invaded the matrigel locally around the mass of the tumor cells (top left). However, when stimulated with IL-17A, MDA-MB231 exhibited increased local invasiveness (bottom left) with much more aggressive features (bottom right, highly invasive extensions indicated by arrows). Furthermore, invasion was not restricted to the cellular mass as the tumor cells were disseminated all around the matrigel and became organized like a network (top right) which is typical of high aggressiveness. In conclusion, IL-17A increases pro-migratory and pro-invasive properties of breast cancer cells and confers an aggressive and highly invasive phenotype in 3D cultures.

IL-17A promotes breast cancer cell migration and invasion.
(A) MCF7 cells were cultured in complete medium alone (medium) or supplemented with 1, 10 or 100 ng/ml of recombinant human IL-17A (IL-17A) as indicated. Cell migration was evaluated in transwell migration assay (upper panel, representative photomicrographs; lower panel, quantification: mean ± SEM of two independent experiments). (B and C) MDA-MB231 were cultured for 2 days in complete medium alone (medium) or supplemented with 100 ng/ml of recombinant human IL-17A as indicated. Cell invasiveness was then evaluated using Matrigel Invasion Chambers (B, upper panel, representative photomicrographs; lower panel, quantification: mean ± SEM of three independent experiments) and using 3D Clusters assays (C, upper left: invasive boarder of untreated cells; lower left and right: invasive boarder of IL-17A-treated cells; upper right: invasion of the surrounding ECM by IL-17A treated cells). (* P < 0.05; ** P < 0.01; *** P < 0.001).
IL-17A released by TILs promotes tumor cell chemoresistance, proliferation and migration
We then asked whether infiltrating T lymphocytes that secrete IL-17A would mediate similar effects on tumor cells as the ones obtained with recombinant IL-17A and whether the effects of TILs could be neutralized by IL-17A blocking monoclonal antibodies. To address this question, we cultured breast cancer cell lines with TIL-conditioned medium. We collected culture supernatant from IL-17A-producing TILs isolated from patient AL (TIL(AL)) and tested whether it could promote resistance to docetaxel, proliferation and migration in an IL-17A-dependent manner. As TILs secrete many soluble factors, the specific contribution of IL-17A was determined using anti-IL-17A neutralizing antibodies or their matched isotype control. Recombinant IL-17A was very potent at protecting BT20 cells from docetaxel-induced cell death (Figure 4) and TIL(AL)-conditioned medium also inhibited docetaxel-induced cytotoxicity (Figure 8A). Specific neutralization of IL-17A using OREG-203 monoclonal antibody markedly decreased the protection mediated by TIL(AL). TIL(AL) also increased the proliferation of T47D cells (which responded well to recombinant IL-17A, Figure 6). Again, this effect was diminished by neutralizing IL-17A with OREG-203 but not by the control antibody (Figure 8B). TIL(AL) also stimulated the migration of MCF7 cells (as observed with the recombinant cytokine, Figure 7A) which was abrogated by antibody-mediated neutralization of IL-17A (Figure 8C). Similar results were obtained with OREG-210, a distinct IL-17A neutralizing monoclonal antibody (data not shown). In aggregates, we found that IL-17A is released by breast cancer TILs and mediates proliferation, chemoresistance (at least in part through the ERK kinases) and migration/invasion. Treatment with IL-17A-neutralizing antibodies abrogated the pro-oncogenic effects of IL–17A released by TILs.

Physiological IL-17A from TIL supernatants promotes tumor cell chemoresistance, proliferation and migration.
(A) BT20 breast cancer cells were cultured in complete medium alone (medium) or supplemented with TIL(AL)-conditioned medium (1/10 vol/vol) and control antibody (control Ab) or IL-17A-neutralizing antibody (OREG-203) at 10 μg/ml as indicated for 48 h. Cells were then switched in FCS-free medium supplemented with TIL(AL)-conditioned medium (1/10 vol/vol) and antibodies as indicated for 24 h and then further treated with docetaxel at 20 or 40 μg/ml for 7 h. The cytotoxicity was determined using the Cytotoxicity Detection Kit (Roche). Data shown are representative of 3 independent experiments. (B) T47D cells were cultured in complete medium supplemented with TIL(AL)-conditioned medium (1/10 vol/vol) and control antibody (control Ab) or IL-17A-neutralizing antibody (OREG-203) at 10 μg/ml as indicated. Cell proliferation was assessed at 72 h using tritiated thymidine ([3H]-TdR) incorporation protocol. Data are representative of 3 independent experiments performed in triplicates. (C) MCF7 cells were cultured in complete medium alone (medium) or supplemented with TIL(AL)-conditioned medium (1/10 vol/vol) and control antibody (control Ab) or IL-17A-neutralizing antibody (OREG-203) at 10 μg/ml as indicated. Cell migration was evaluated in transwell migration assay. Data are the mean ± SEM of two independent experiments, each performed in triplicates. (* P < 0.05; ** P < 0.01; *** P < 0.001).
Discussion
We demonstrated here that IL-17A producing cells infiltrate breast cancer. Our results from human tissue microarrays and human biopsies confirmed previous observations6,8,9,10 reporting upregulation of IL–17A or IL-17A producing cells in breast cancers. Some tumors were infiltrated with high number of IL–17A positive immune cells, whereas no or few IL-17A positive cells were observed in the stroma of healthy mammary tissues. We observed that most IL-17A+ immune cells were lymphocytes and macrophages, which is in accordance with previous reports6. In silico analyses of a published study from reference19 using the ONCOMINE software sustain the conclusions that IL17A mRNA is increased in breast cancer stroma (P < 10−6) compared to the stroma of normal breast. In this dataset, IL17A expression is higher in the tumors that recurred versus the non-recurrent diseases and higher in the poor prognosis group versus the good prognosis group.
In vitro experiments presented here, using cell lines and primary material from cancer patients, suggest that IL-17A may promote breast cancer cell proliferation, invasion as well as resistance to chemotherapy. Therefore, IL-17A is suspected to be a poor prognosis factor in breast cancer. Our breast cancer cohort (SUPER BIO CHIPS, slide CBA3) contains a small number of cancer cases with limited clinical information, which did not allow us to investigate the clinical relevance of IL-17A upregulation in breast cancer or perform statistical analyses. However, patients with IL-17A positive tumors tend to have a higher frequency of lymph node metastasis (75% of IL-17A+ tumors are N+ compared to 63% of IL-17A- tumors that are N+) which may reflect the fact that IL-17A produced by TILs stimulates breast cancer cell migration and invasion as reported by us (Figures 7 and 8C) and others6. We cannot conclude regarding its role in systemic metastases, resistance to therapy or its overall impact on survival.
To gain insight into the clinical relevance of IL-17A in breast cancer we would like to refer to the study published recently by Chen and colleagues10. To our knowledge, this is the only study assessing the expression of IL-17A by IHC in a large number of breast cancer patients (207 breast cancer cases) with extensive clinical parameters. In this cohort, infiltration of breast cancer by IL-17A producing cells significantly correlates with higher tumor grade (P < 0.01), ER and PR negative status (P < 0.01) and triple negative tumors (P < 0.05). Patients with IL-17A positive tumors have shorter DFS compared to IL-17A negative tumors (5-year DFS of 64% compared to 87.3%, respectively, P < 0.01). In univariate Cox analysis, elevated IL-17A is a significant prognostic factor influencing DFS (P < 0.01). Multivariate Cox proportional hazard analysis also showed that elevated IL-17A was a significant prognostic factors for DFS after controlling for age, histology, tumor grade and nodal status and expression status of ER and PR (P < 0.05).
One of the main finding of our work is that IL-17A derived from breast cancer TILs may influence the response and outcome of chemotherapy treatment. Again, the above mentioned study seems to support our conclusion. Among their 207 breast cancer patients, 58 presented with locally advanced breast cancer and received neoadjuvante chemotherapy. Although IL-17A was not statistically associated with the response to chemotherapy, the presence or absence of IL-17A drastically influenced the DFS of patients receiving chemotherapy (P < 0.01) as 5-year DFS dropped from 81.5% for IL-17A- patients to 38.5% for IL-17A+ patients. Because the impact of IL-17A on DFS is much stronger in the context of chemotherapy (81.5% compared to 38.5%) than in the entire cohort (87% to 64%), we believe that this points out a particular impact of IL-17A within this subgroup of patients with locally advanced tumors receiving chemotherapy. In aggregate, the clinical data form the 207 breast cancer patient cohort of Chen et al. strongly supports the notion that IL-17A is a poor prognostic factor in breast cancer and is associated with ER- or triple negative, high grade tumors with shortened DFS.
Our and other10 studies raise the conclusion that IL-17A is involved in ER(−) tumors, including triple negative tumors. The mechanisms underlying the association between IL-17A and ER deficiency remained unknown. However, it is interesting to mention that several studies reported a direct relation between estrogen status and IL-17A20,21 and estrogen/ER signaling deficiency was shown to promote the differentiation of Th17 cells21. Therefore, one could speculate that ER deficiency in breast tumors may directly promote the expansion of IL-17A+ TILs, a possibility that would require further investigation. We believe that this speculation is a plausible scenario as we demonstrated here that ER(+) and ER(−) cell lines respond to IL-17A in a similar manner. Therefore, the association of ER deficiency and IL-17A expression may not reflect a functional synergy but rather an inter-regulation or co-regulation by a common pathway.
We showed here that IL-17A exerts several oncogenic effects on cancer cells including resistance to chemotherapy-induced cell death, proliferation, migration and invasion. The modulation of drug sensitivity of cancer cells by IL-17A is an important oncogenic property of IL-17A with clinical significance and it has never been reported. However, it was found that IL-17A promotes survival of synoviocytes in rheumatoid arthritis through activation of STAT3-Bcl-2 axis22 and confers broad chemoresistance to dendritic cells through the regulation of the Bcl-2 family member BCL2A123. Using knockout (KO) mice, Yu and colleagues demonstrated that IL-17A controls tumor progression by regulating the STAT3- Bcl-2/Bcl-XL pathway in several cancer models24,25. Here we report that IL-17A activates the ERK pathway, an important survival pathway for cancer cells and we demonstrated that this pathway is a crucial mediator of IL-17A-induced resistance to docetaxel. Altogether these results suggest that IL-17A present in the tumor microenvironment may be an important survival factor and source of therapy resistance for breast cancer cells. We also evidenced that IL-17A promotes the proliferation of some breast cancer cell lines. Although this had not been reported previously for cancer cells, it had been demonstrated that IL-17A promotes the proliferation of smooth muscle airway cells14 and human mesenchymal stem cells26 through the activation of the MEK-ERK pathway. However, IL-17A did not systematically increase the proliferation of breast cancer cell lines, at least in vitro. This may reflect the fact that the cell lines used have been kept in Fetal Calf Serum (FCS) rich-medium for a long period and their proliferation is unlikely to rely on external factors. We are currently testing the effect of IL-17A on primary cells or freshly isolated tumor cells, models that may be more relevant to assess its impact on proliferation. To our knowledge, this is the first report that used primary material from cancer biopsies to assess in depth the role of IL-17A released by TILs from breast cancer patients. Importantly, these freshly isolated TILs exert similar pathophysiologic effects as described above mainly through IL-17A as specific neutralization of IL-17A largely abrogated their effects. These observations are critical to understand the role of IL-17A in breast carcinogenesis, as TILs secrete many soluble factors that could contribute to their pro-tumor effects.
Whereas IL-17A or IL-17A producing immune cells are unambiguously increased in numerous tumors including breast cancers, their pro- versus anti-tumor effects remains debated27,28. When signaling on the tumor cells or the tumor vasculature, IL-17A may favor tumor progression by promoting cell proliferation, survival, chemoresistance and dissemination as well as angiogenesis29,30,31. On the other hand Th17 were demonstrated to exert antitumor activity32,33. Interestingly, these two studies demonstrated that the antitumor response mediated by Th17 cells was independent of IL-17A and rather relied on IFN–γ secretion. Therefore, as previously suggested by Maniati and colleagues in their review28, we believe that our work could indeed help to reconcile the pro and antitumor effects of IL-17A and Th17 cells as IL–17A may favor tumor progression whereas Th17 cells may exert antitumor effects mainly through IFN–γ production. In line with such speculation, the specific role of IL-17A, independently of the cells that produce it, can be addressed in IL17A KO mice. These mice consistently exhibit a tumor-resistant phenotype. They have reduced development of TPA/DMBA-induced skin tumors24, decreased colon tumor initiation in the APC+/− genetic background34 and exhibited reduced growth of the B16 melanoma and MB49 bladder carcinoma cells25. IL17A KO mice also exhibited reduced growth, decreased metastasis and improved survival when challenged with lung cancer cell lines17,35. Along similar lines, blockade of IL-17A signaling in IL17RC KO mice revealed decreased tumor progression in a prostate cancer model36. Furthermore, anti-IL-17A antibody therapy decreased tumor growth and metastasis in colon cancer31 and breast cancer37,38 mouse models. Therefore, the role allotted to IL-17A itself seems to be tumor and metastasis-promoting. This suggests an interesting therapeutic opportunity where IL-17A neutralization could block pro-tumor and pro-metastatic effects of IL-17A while allowing Th17-mediated tumor eradication through IFN–γ production.
Methods
Cell culture and reagents
MCF7, T47D, BT20, MDA-MB468, MDA-MB157 and MDA-MB231 cell lines were obtained from the American Type Culture Collection. MCF7, T47D, BT20 and MDA-MB231 were cultured in RPMI medium (Invitrogen) supplemented with 10% Fetal Calf Serum (FCS), 2% glutamine and 1% antibiotics (Life technologies). MDA-MB468 cell line was cultured in DMEM/F12 medium (Life Technologies) supplemented as described above. MDA-MB157 cell line was cultured in L15/DMEM medium (Life Technologies) supplemented as described above. All cells were kept at 37°C in 5% CO2 atmosphere incubator. Recombinant human IL-17A was purchased at Peprotech (Rocky Hill, NJ).
IL-17A immunohistochemistry
IL-17A staining on tissues arrays (SUPER BIO CHIPS, slide CBA3) was performed by a pathologist using a standard peroxidase method according to a protocol adapted from Coury et al39. and validated on renal allograft paraffin sections from patient with chronic active rejection. Goat polyclonal anti-human IL-17A antibody (dilution 1:40, R&D Systems, Minneapolis, MN) was employed as the primary antibody followed by anti-goat antibody (LSAB kit, DAKO). IL-17A expression was scored −, +, ++ and +++ for the absolute number of IL-17A positive immune cells within the stroma (see Supplementary Figure 1).
Clinical breast cancer biopsies
Human biopsy samples were obtained from the cancer center Institut Jean Godinot. This study was made according the approval of an ethic committee and all patients were informed and agreed the study.
Tumor-infiltrating lymphocyte (TIL) cultures
TILs from 6 ER(−) breast cancer biopsies were isolated and expanded according to the protocol adapted from Bagot M et al40. Briefly, TILs from patients were obtained from surgical tumor fragments mechanically dispersed into single-cell suspensions and cultured in 12-well plates with culture medium consisting of RPMI 1640, 2 mmol/L L-glutamine, penicillin (100 U/mL), streptomycin (100 μg/mL), 10% heat-inactivated human serum and 100 U/mL interleukin-2. Cultures were then fed every 3 days with IL-2–containing culture medium and the growing wells were expanded for 2 weeks. TILs were then washed and cultured in IL-2–containing medium with irradiated PBL (feeders) stimulated with PHA at 1 μg/ml. After 3 days the expanded TILs were extensively washed and fed with IL-2–containing medium for 6 days. Cells were harvested for IL-17A secretion and stained with anti-CD3, anti-CD4, anti-CD8 and anti-TCRγδ antibodies for flow cytometry analysis. Production of IL-17A by TILs was measured by ELISA in cell culture supernatants of 16 h cultures in PHA/IL-2 activation medium.
Quantification of IL-17A in cell culture supernatants
IL-17A from cell culture supernatants was quantified using commercial ELISA kit (R&D Systems) following instructions of the manufacturer.
Isolation of mRNA and quantitive RT-PCR
Total RNA was isolated using the GenElute™ Mammalian Total RNA kit (Sigma–Aldrich, St. Louis, MO) following the manufacturer's instructions. Total RNA (1 μg) was treated with 1 U/μg RNA of DNase I Amplification Grade (Invitrogen) according to the manufacturer's instructions and in the presence of 10 U/μg RNA of RNaseOUT (Invitrogen). After DNase inactivation, RNA was reverse transcribed using random nonamers (Promega, Madison, WI) and M-MLV Reverse Transcriptase H Minus (Promega) according to the manufacturer's instruction.
The forward and reverse primers used in the PCR reaction were designed with Primer-BLAST software except for IL-17A (described in ref41) and GAPDH (described in ref42) and were as follow: GAPDH-forward 5′-GAAGGTGAAGGTCGGAGTCA-3′, GAPDH-reverse 5′-GACAAGCTTCCCGTTCTCAG-3′, IL-17A-forward 5′- ACTACAACCGATCCACCTCAC-3′, IL-17A-reverse 5′- ACTTTGCCTCCCAGATCACAG-3′, IL-17 RA-forward 5′-TGCCCCTGTGGGTGTACTGGT-3′, IL-17 RA-reverse 5′-GCAGGCAGGCCATCGGTGTA-3′, IL-17 RC-forward 5′-GGCTTGGTTTCACGCGCAGC-3′ and IL-17 RC-reverse 5′-CGGCCCTGCAAGAAGTCGGG-3′.
Real-time polymerase chain reaction quantification was carried out with the LightCycler 480 II System (Roche Diagnostics, Basel, Switzerland) using the SYBR Premix Ex Taq (Tli RNaseH Plus) kit (Ozyme, Saint-Quentin-en-Yvelines, France). The cycling conditions were as follows: 95°C for 1 min followed by 40 cycles of 95°C for 20 s, 60°C for 20 s and 72°C for 30 s. The sizes of the RT-PCR products were confirmed by agarose electrophoresis. At the end of the amplification, a melting temperature analysis of the amplified gene products was performed routinely for all cases. The PCR products were melted by gradually increasing the temperature from 60 to 95°C in 0.3°C steps and the dissociation curves were generated with the Melting Curve analysis tool of the LightCycler 480 software (Roche Diagnostics). We confirmed that only one product was consistently amplified in all PCR reactions. The negative water control showed no amplification.
ERK1/2 immunoblotting
Cells were seeded at 3.106 cells per well in 6-well plates and cultured overnight in FCS-free medium. Cells were stimulated with IL-17A at 10 ng/ml and/or U0126 at 10 μM for 20, 30 min or 3 h as indicated. The medium was then removed, the cells were lysed in 1% Triton X100 lysis buffer, incubated for 1 h on ice and centrifuged at 4°C for 20 min at 10,000 g. The supernatants were collected and protein concentration was determined using the Bradford Assay (Bio-Rad, Hercules, CA). Proteins (70 μg) were resolved in 8% SDS–PAGE and transferred on nitrocellulose membrane. The membrane was blocked for 1 h at room temperature, by using 5% nonfat milk in tris-buffered saline (TBS) containing 0.1% Tween 20 (Sigma–Aldrich) and incubated overnight at 4°C with a monoclonal rabbit anti-phospho-p44/42 MAPK (Erk1/2) antibody (1:1,000), a monoclonal rabbit anti-p44/42 MAPK (Erk1/2) antibody (1/1,000) or with a monoclonal mouse anti-actin antibody (1:1,000) (Cell Signalling, Danvers, MA) used as a loading control, in blocking solution. After 3 washes, the membrane was incubated 1 h at room temperature with goat anti-rabbit or goat anti-mouse secondary antibodies (1:10,000) (Jackson Immunoresearch, West Grove, PA) conjugated to horseradish peroxydase.
Cytotoxicity assay
MCF7, T47D (1000 cells/well), BT20, MDA-MB468 and MDA-MB157 (3,000 cells/well) cells were seeded in a 96 wells plate in adequate complete medium alone or treated with recombinant human IL-17A (1 or 10 ng/ml) or TIL(AL)-conditioned medium (1/10 vol/vol) and/or antibodies (10 μg/ml) as indicated. After 48 h of culture, medium was changed to a FCS-free one supplemented with corresponding concentration of cytokines or TIL(AL)-conditioned medium and/or antibodies and further supplemented with U0126 inhibitor (10 μM) when indicated. After 24 h, culture medium is then further supplemented with docetaxel at 5, 10, 20 or 40 μg/ml as indicated. Untreated cells (control medium) and Triton X100 treated cells (100% cell death) were used as controls. Each condition was performed in duplicates. The cytotoxicity was determined using the Cytotoxicity Detection Kit (Roche) according to the manufacturer's instructions. To this aim, 50 μl of supernatant from each well were collected into a 96 wells plate and incubated with 50 μl of freshly prepared Reaction Mixture for 30 minutes at room temperature. Optical density was then read at 490 nm. The percentage of cytotoxicity is calculated as followed: % = 100 × (exp value - control medium value)/(Triton X100 treated cells value - control medium value).
Cell proliferation assay ([3H]-TdR)
MCF7, T47D, MDA-MB468 (500 cells/well) and BT20 (1,000 cells/well) were plated on 96-well plates and maintained for 24 h in complete medium. Medium is then removed and replaced with complete medium supplemented with recombinant human IL-17A (0, 1, 10 or 100 ng/ml) or TIL(AL)-conditioned medium (1/10 vol/vol) and/or antibodies (10 μg/ml) as indicated. After 72 h of culture, cells were pulsed with 1 μCi of tritiated thymidine ([3H]-TdR) per well. [3H]-TdR uptake was measured using PerkinElmer PerkinElmer MicroBeta2 plate counter (PerkinElmer, Waltham, MA, USA). Each experiment was performed in hexaplicates.
Transwell migration assay
2.104 cells were seeded on the upper compartment of transwell chambers (24-well insert; pore size, 8 μm; BD Biosciences) in 1% FCS medium alone (medium) supplemented with recombinant human IL-17A (1, 10 or 100 ng/ml) or TIL(AL)-conditioned medium (1/10 vol/vol) and/or antibodies (10μg/ml) for 22 hours at 37°C as indicated. Medium supplemented with 10% FCS is added to the lower compartment of the chambers. The cells on the transwell were stained with 0.5% crystal violet prior imaging and enumeration. Total number of migrating cells was calculated by analyzing 5 fields per well in at least two independent experiments performed in duplicates, that is 10 fields per condition.
Matrigel invasion assay
MDA-MB231 cells were cultured in complete medium supplemented with 100 ng/ml of recombinant IL-17A for 2 days. Matrigel Invasion Chambers (BD BioCoat™ BD Matrigel™ Invasion Chamber, 24-well Cell culture inserts) were then used to study the invasiveness of cancer cells. 2.104 cells are then added to the upper compartment of the chambers in 1% FCS medium alone or supplemented with 100 ng/ml of recombinant cytokines as indicated. Medium supplemented with 10% FCS is added to the lower compartment of the chambers. Plates are incubated for 22 hours at 37°C. Transwell filters are then fixed and stained in Giemsa solution following which non-invading cells are removed from the upper surface of the transwell membrane using a cotton swab. Images of cells from three representative fields are captured digitally and the number of cells present on the transwell is counted. Data are mean of three independent experiments performed in triplicates.
3D Cluster assays
MDA-MB231 cells were cultured in complete medium supplemented with 100 ng/ml of recombinant IL-17A for 2 days. 275 μl of cold Basement Membrane Matrix (LDEV-Free, 354234, BD Biosciences) was added per refrigerated CultureSlides (8-wells, 354118, BD Biosciences). Inserts were then incubated for 20 min at 37°C to obtain a solid plug of Matrigel. 106 pretreated cells were centrifuged and 1 μl of the cell pellet was loaded directly into the plug of Matrigel. 10 μl of Matrigel was added on top of the plug and inserts were incubated at 37°C for 5 min. 250 μl of culture medium supplemented or not with the cytokines was added on top and inserts were kept at 37°C, 5% CO2 humidified incubator and invasive behavior was analyzed at 24 h.
Statistical analyses
All values are expressed as mean +/− SEM unless otherwise specified. Quantitative data were compared using student's t test. The dose-dependent effects on cell proliferation (Figure 6) and migration (Figure 7A) were analyzed using the Kruskal-Wallis test. P < 0.05 was considered significant.
References
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444, 10.1038/nature07205 (2008).
Gaffen, S. L. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 9, 556–567, 10.1038/nri2586 (2009).
Onishi, R. M. & Gaffen, S. L. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129, 311–321, 10.1111/j.1365-2567.2009.03240.x (2010).
Iwakura, Y., Ishigame, H., Saijo, S. & Nakae, S. Functional specialization of interleukin-17 family members. Immunity 34, 149–162, 10.1016/j.immuni.2011.02.012 (2011).
Ji, Y. & Zhang, W. Th17 cells: positive or negative role in tumor? Cancer Immunol Immunother 59, 979–987, 10.1007/s00262-010-0849-6 (2010).
Zhu, X. et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res 10, R95, 10.1186/bcr2195 (2008).
Jung, M. Y., Kim, S. H., Cho, D. & Kim, T. S. Analysis of the expression profiles of cytokines and cytokine-related genes during the progression of breast cancer growth in mice. Oncol Rep 22, 1141–1147 (2009).
Su, X. et al. Tumor microenvironments direct the recruitment and expansion of human Th17 cells. J Immunol 184, 1630–1641, 10.4049/jimmunol.0902813 (2010).
Benevides, L. et al. Enrichment of regulatory T cells in invasive breast tumor correlates with the upregulation of IL-17A expression and invasiveness of the tumor. Eur J Immunol, 10.1002/eji.201242951 (2013).
Chen, W. C. et al. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology, 10.1111/his.12156 (2013).
Nam, J. S. et al. Transforming growth factor beta subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res 68, 3915–3923, 10.1158/0008-5472.CAN-08-0206 (2008).
MacKeigan, J. P., Collins, T. S. & Ting, J. P. MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem 275, 38953–38956, 10.1074/jbc.C000684200 (2000).
Favata, M. F. et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273, 18623–18632 (1998).
Chang, Y. et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. Faseb J 26, 5152–5160, 10.1096/fj.12-208033 (2012).
Acosta, J. J. et al. Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol Endocrinol 17, 2268–2282, doi:10.1210/me.2002-0422 (2003).
Gu, F. M. et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer 10, 150, 10.1186/1476-4598-10-150 (2011).
Li, Q. et al. IL-17 promoted metastasis of non-small-cell lung cancer cells. Immunol Lett 148, 144–150, 10.1016/j.imlet.2012.10.011 (2012).
Li, J. et al. Interleukin 17A promotes hepatocellular carcinoma metastasis via NF-kB induced matrix metalloproteinases 2 and 9 expression. PLoS One 6, e21816, 10.1371/journal.pone.0021816 (2011).
Finak, G. et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14, 518–527, 10.1038/nm1764 (2008).
DeSelm, C. J. et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem 113, 2895–2902, 10.1002/jcb.24165 (2012).
Tyagi, A. M. et al. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One 7, e44552, 10.1371/journal.pone.0044552 (2012).
Lee, S. Y. et al. IL-17-mediated Bcl-2 expression regulates survival of fibroblast-like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Res Ther 15, R31, 10.1186/ar4179 (2013).
Olsson Akefeldt, S. et al. Chemoresistance of human monocyte-derived dendritic cells is regulated by IL-17A. PLoS One 8, e56865, 10.1371/journal.pone.0056865 (2013).
Wang, L., Yi, T., Zhang, W., Pardoll, D. M. & Yu, H. IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res 70, 10112–10120, 10.1158/0008-5472.CAN-10-0775 (2010).
Wang, L. et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 206, 1457–1464, 10.1084/jem.20090207 (2009).
Huang, H. et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 16, 1332–1343, 10.1038/cdd.2009.74 (2009).
Hemdan, N. Y. Anti-cancer versus cancer-promoting effects of the interleukin-17-producing T helper cells. Immunol Lett 149, 123–133, 10.1016/j.imlet.2012.11.002 (2013).
Maniati, E., Soper, R. & Hagemann, T. Up for Mischief? IL-17/Th17 in the tumour microenvironment. Oncogene 29, 5653–5662, 10.1038/onc.2010.367 (2010).
Numasaki, M. et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood 101, 2620–2627, 10.1182/blood-2002-05-1461 (2003).
Numasaki, M. et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 175, 6177–6189 (2005).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15, 1016–1022, 10.1038/nm.2015 (2009).
Muranski, P. et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112, 362–373, 10.1182/blood-2007-11-120998 (2008).
Hinrichs, C. S. et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood 114, 596–599, 10.1182/blood-2009-02-203935 (2009).
Chae, W. J. et al. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci U S A 107, 5540–5544, 10.1073/pnas.0912675107 (2010).
Carmi, Y. et al. Microenvironment-derived IL-1 and IL-17 interact in the control of lung metastasis. J Immunol 186, 3462–3471, 10.4049/jimmunol.1002901 (2011).
Zhang, Q. et al. Interleukin-17 promotes formation and growth of prostate adenocarcinoma in mouse models. Cancer Res 72, 2589–2599, 10.1158/0008-5472.CAN-11-3795 (2012).
Das Roy, L. et al. Breast-cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res 11, R56, 10.1186/bcr2345 (2009).
Novitskiy, S. V. et al. TGF-beta receptor II loss promotes mammary carcinoma progression by Th17 dependent mechanisms. Cancer Discov 1, 430–441, 10.1158/2159-8290.CD-11-0100 (2011).
Coury, F. et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med 14, 81–87, 10.1038/nm1694 (2008).
Bagot, M. et al. Isolation of tumor-specific cytotoxic CD4+ and CD4+CD8dim+ T-cell clones infiltrating a cutaneous T-cell lymphoma. Blood 91, 4331–4341 (1998).
Miyagaki, T. et al. IL-22, but not IL-17, dominant environment in cutaneous T-cell lymphoma. Clin Cancer Res 17, 7529–7538, 10.1158/1078-0432.CCR-11-1192 (2011).
Mayer, C. et al. DNA repair capacity after gamma-irradiation and expression profiles of DNA repair genes in resting and proliferating human peripheral blood lymphocytes. DNA Repair (Amst) 1, 237–250 (2002).
Acknowledgements
The authors wish to thank F. Dijoud for performing IHC on breast cancer TMA. This work is supported by grants from INSERM, Agence Nationale pour la Recherche and OREGA Biotech.
Author information
Authors and Affiliations
Contributions
J.B. and A.B. designed and supervised the study and wrote the main manuscript text. S.C., J.G., C.T., E.L. performed experiments and prepared the figures. C.G., A.M.S., H.C., C.M. provided biopsies. G.A., N.B., J.F.E. contributed to the design and interpretation of experiments and to the editing of the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
Drs Bonnefoy, Bensussan, Alberici and Eliaou are cofounders and shareholders of OREGA Biotech. Dr Bastid is an employee of OREGA Biotech. Drs Cochaud, Giustiniani, Thomas, Laprevotte, Garbar, Savoye, Curé and Mascaux declare no conflict of interest.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cochaud, S., Giustiniani, J., Thomas, C. et al. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci Rep 3, 3456 (2013). https://doi.org/10.1038/srep03456
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03456
This article is cited by
-
The IL-17 family in diseases: from bench to bedside
Signal Transduction and Targeted Therapy (2023)
-
Identifying drivers of breast cancer metastasis in progressively invasive subpopulations of zebrafish-xenografted MDA-MB-231
Molecular Biomedicine (2022)
-
Sestrin2 in cancer: a foe or a friend?
Biomarker Research (2022)
-
Innovative targets of the lncRNA-miR-mRNA network in response to low-dose aspirin in breast cancer patients
Scientific Reports (2022)
-
Single nucleotide variants in immune-response genes and the tumor microenvironment composition predict progression of mantle cell lymphoma
BMC Cancer (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
