
Abstract
Hematopoietic stem and progenitor cells (HPCs) can be maintained in vitro, but the vast majority of their progeny loses stemness during culture. In this study, we compared DNA-methylation (DNAm) profiles of freshly isolated and culture-expanded HPCs. Culture conditions of CD34+ cells - either with or without mesenchymal stromal cells (MSCs) - had relatively little impact on DNAm, although proliferation is greatly increased by stromal support. However, all cultured HPCs - even those which remained CD34+ - acquired significant DNA-hypermethylation. DNA-hypermethylation occurred particularly in up-stream promoter regions, shore-regions of CpG islands, binding sites for PU.1, HOXA5 and RUNX1 and it was reflected in differential gene expression and variant transcripts of DNMT3A. Low concentrations of DNAm inhibitors slightly increased the frequency of colony-forming unit initiating cells. Our results demonstrate that HPCs acquire DNA-hypermethylation at specific sites in the genome which is relevant for the rapid loss of stemness during in vitro manipulation.
Similar content being viewed by others


Introduction
Culture expansion of hematopoietic stem and progenitor cells (HPCs) remains one of the major challenges in stem cell biology. This is of particular relevance for umbilical cord blood (CB) where the number of available stem cells is limited and often insufficient for transplantation of adult patients1. It is also crucial for in vitro manipulation of HPCs in gene therapy approaches2. Proliferation of HPCs can be stimulated in vitro but it is usually associated with loss of stem cell properties. Culture conditions can be optimized by combinations of growth factors3, small synthetic chemicals4 or specific proteins including Notch-ligands5. It has also been shown that co-culture with mesenchymal stromal cells (MSCs) mimics components of the hematopoietic niche and thereby supports maintenance of primitive HPCs in vitro6,7,8. However, all of these approaches have in common that the vast majority of the proliferating progeny loses regenerative potential within few days. A better understanding of molecular changes during culture might facilitate controlled in vitro expansion of the multipotent subset.
DNA-methylation (DNAm) of CpG dinucleotides is a key epigenetic modification. Upon cell division, the DNAm pattern is maintained on the newly synthesized DNA strand particularly by DNA methytransferase 1 (DNMT1), whereas DNMT3A and DNMT3B act as de novo methyltransferases and modify unmethylated CpG sites during differentiation9. Conversely, active demethylation may be promoted by methyl-CpG binding proteins or via a hydroxymethylate intermediate step10. DNAm plays a central role in normal hematopoietic development and consequently, it might also be relevant for the rapid loss of stem cell activity during in vitro culture11.
In this study, we analyzed if the DNAm pattern of CB derived CD34+ HPCs is modified during culture expansion either with or without stromal support. DNAm profiles were determined using a novel Infinium HumanMethylation450 platform which assays more than 480,000 CpG sites at single base resolution (covering 99% of RefSeq genes and 96% of CpG islands)12. We demonstrate that culture expansion induces specific hypermethylation in relevant hematopoietic genes.
Results
Expansion of hematopoietic progenitor cells affects DNAm profiles
CD34+ cells were cultured for seven days either on tissue culture plastic (TCP) or in co-culture with MSCs (Fig. 1a). Notably, the CD34+ fraction is heterogeneous and only a small subset resembles hematopoietic stem cells (HSCs). Stromal support greatly increased cellular proliferation, the percentage of CD34+ cells and colony forming unit (CFU)-frequency (Fig. 1b–d). We have previously shown that these culture conditions expand CD34+ cells in vitro and that NOD/SCID mice repopulating cells were particularly maintained with stromal support13. The cellular size of HPCs increased during culture, they became more elongated and overall CD34 and CD133 expression decreased, whereas differentiation markers were upregulated (Supplementary Fig. S1). Taken together, stromal support facilitates expansion of HPCs, but the vast majority of the progeny further proceeds towards differentiation.

Culture expansion of CD34+ cells affects DNAm.
(a) Experimental design. CD34+ cells (d0) were cultured in parallel either with or without mesenchymal stromal cells (MSCs). After one week, HPCs were separated into CD34+ and CD34− fractions. (b) MSCs significantly stimulated proliferation and maintenance of CD34 expression. (c) Colony-forming unit (CFU)-frequency of freshly isolated and expanded HPCs. CFU-initiating cells were particularly increased by stromal support of MSCs (n = 3; *p ≤ 0.05; **p ≤ 0.01). (d) HPCs were stained with carboxyfluorescein diacetate N-succinimidyl ester (CFSE) before culture expansion. As indicated in the histograms, each peak is associated with a unique number of cell divisions. MSCs enhance proliferation of HPCs and maintain CD34 expression for more cell divisions. (e) Scatter plots demonstrate significant DNAm changes upon culture expansion (adjusted p < 0.05). In comparison to freshly isolated CD34+ (d0) cells, all expanded fractions (CD34+ w/o MSC, CD34− w/o MSC or CD34+ w/MSC) revealed particularly hypermethylation at specific CpG sites. (f) Number of differentially methylated CpG sites (hyper- and hypomethylated) between the different subsets. DNAm profiles of CD34+ w/o MSC and CD34+ w/MSC were closely related. (g) DNAm of CpG sites is exemplarily depicted for the genes CD34, CD133 and GATA1. Promoter regions of CD34 and CD133 become higher methylated in the CD34− progeny. Conversely, GATA1 is rather hypomethylated and up-regulated on gene expression level. Significantly changed CpG sites (adjusted p < 0.05) are indicated by arrowheads (vs. CD34+ (d0) depicted in green; vs. CD34− w/o MSC in blue; *p ≤ 0.05; **p ≤ 0.01). See also Supplementary Fig. S1 and S2.
We expected that DNAm changes were particularly acquired in the faster proliferating subset which loses CD34 expression. To test this thesis, we separated cultured HPCs into a CD34+ and a CD34− fraction and compared their DNAm profiles with freshly isolated HPCs. Furthermore, HPCs were co-cultured with MSCs to estimate the impact of stromal support on DNAm profiles. CD34− w/MSCs were excluded due to contamination of MSCs. The overall DNAm level was hardly affected by culture expansion (Supplementary Fig. S2a,b), but there were many significant changes at specific CpG sites (adjusted p-value < 0.05): CD34+ cells without stromal support (CD34+ w/o MSC) accumulated 15,271 hypermethylated and 890 hypomethylated CpG sites; CD34− w/o MSC cells showed 17,140 hypermethylated and 4,073 CpG hypomethylated sites; and the CD34+ w/MSC fraction revealed 15,668 hypermethylated and 2,519 hypomethylated CpG sites (Fig. 1e). Thus, in vitro culture of HPCs results predominantly in hypermethylation of specific CpG sites.
Unexpectedly, DNAm profiles of culture expanded CD34+ versus CD34− subsets revealed fewer differences: 4,304 CpG sites were higher methylated in CD34+ w/o MSC, whereas 1,864 CpG sites were higher methylated in CD34− w/o MSC (Fig. 1f, Supplementary Fig. S2c). We reasoned that these DNAm changes might reflect differentiation of the CD34− subset. In fact, some of the most significant hypermethylation in CD34− w/o MSC was observed in CD34 (adjusted p = 0.0003) and CD133 (p = 0.005), whereas several genes involved in hematopoietic differentiation, such as GATA binding protein 1 (GATA1; p = 0.0007), were rather hypomethylated (Fig. 1g). To validate DNAm changes we have exemplarily analyzed the differentially methylated region in the promoter region of the gene CD34 using bisulfite pyrosequencing in independent samples. As observed by the HumanMethylation450 platform the CD34− cell fraction revealed significant hypermethylation (Supplementary Fig. S3a).
Stromal support had even less impact on DNAm profiles: comparison of CD34+ w/o MSC versus CD34+ w/MSC revealed only 848 hypermethylated and 1,116 CpGs hypomethylated CpG sites (Supplementary Fig. S2c). Thus, co-culture with MSCs does not prevent in vitro culture associated DNAm changes, but it seems to shift this process to higher cell division numbers.
DNAm changes are enriched in genes involved in hematopoietic development
Subsequently, we focused on the CpG sites which were differentially methylated upon culture expansion. These modifications might be related to senescence. Long term culture of other cell types, such as MSCs, has been associated with specific senescence-associated DNAm (SA-DNAm) changes, which can be used for monitoring of senescence14,15. However, DNAm changes upon culture of HPCs revealed only a very moderate association with SA-DNAm changes indicating that they were not related to replicative senescence (Supplementary Fig. S2d).
All expanded cell fractions (CD34+ w/o MSC, CD34− w/o MSC and CD34+ w/MSC) revealed a remarkable overlap in hypermethylation (Fig. 2a). Among these was the Wilms tumor 1 gene (WT1; P < 10−123); NOTCH1 (p < 10−25), a known modulator of lineage-specific events in hematopoiesis16; and various genes of the homeobox gene cluster A (particularly HOXA5: p < 10−35; Fig. 2b, Supplementary Fig. S4a). Hypermethylation was also observed in the HOXB cluster (particularly HOXB3; p < 10−17), but not in HOXB4 which has previously been implicated in in vitro expansion of HPCs (Supplementary Fig. S4b)17,18. Other relevant genes with hypermethylated CpG sites include the myeloid translocation gene 16 (MTG16; also known as CBFA2T3; p < 10−150) that has been implicated in the maintenance of stem cell quiescence; the retinoid X receptor alpha (RXRA; p < 10−91), whose down-regulation is essential for neutrophil development and the adenosine a2a receptor (ADORA2A; p < 10−109), which inhibits neutrophil degranulation; and the epigenetic regulators histone deacetylase 9 (HDAC9; p < 10−4; Supplementary Fig. S4c) and DNMT3A (p < 10−46). The highly significant hypermethylation within DNMT3A was further validated by bisulfite pyrosequencing in independent samples (Supplementary Fig. S3b). On the other hand, hypomethylated CpG sites were related to the transcription factor PAX5 (p < 10−5) and recombination activating gene 2 (RAG2; p < 10−8), which are involved in lymphoid differentiation (Supplementary Fig. S4d). Gene Ontology (GO) analysis of hypermethylated genes revealed highly significant enrichment in functional categories of the immune system, hematopoietic development and activation (Fig. 2c), whereas classification of hypomethylated CpG sites was hardly significant and rather associated with signaling.

Distribution of differentially methylated CpG sites.
(a) Overlap of hypermethylated and hypomethylated CpG sites in the expanded cell fractions. (b) For the genes WT1, HOXA5 and NOTCH1 site specific DNAm changes (arrowheads indicate significant CpG sites in comparison to either CD34+ (d0) [green] or CD34− w/o MSC [blue]) and gene expression changes are exemplarily depicted (*p ≤ 0.05; **p ≤ 0.01). (c) Gene Ontology analysis revealed most significant enrichment of DNAm changes in genes related to categories of hematopoietic activation or immune processes (same colour code as in B). (d) Enrichment of DNAm changes in relation to gene regions or CpG islands. Hypermethylation was enriched in TSS1500 and shore regions, whereas hypomethylation was enriched in 3′UTR, intergenic areas and shelf regions. (hypergeometric distribution: +p < 10−5; #p < 10−10; Δp < 10−15).
DNAm changes were then analyzed in the context of gene regions or CpG islands (CGIs; Fig. 2d). Culture associated DNA-hypermethylation was highly significantly enriched in upstream promoter-regions (TSS1500) and in 5′untranslated regions (UTRs). Furthermore, it was enriched in the 2 kb flanking regions of CGIs which are termed shore regions and which are strongly related to gene expression19. In contrast, hypomethylated CpG sites were rather enriched in 3′UTRs, intergenic regions and more distant to CGIs. These results may indicate that particularly DNA-hypermethylation is functionally relevant.
De novo motif discovery in the direct vicinity of hypermethylated CpG sites (124 bp) identified binding sites for forkhead box proteins, HOXA5, PU.1 and RUNX1 amongst several other transcription factors (TFs; Fig. 3a). Alternatively, we scanned for known TF-binding sites and the most significant results were observed for the TFs PU.1 (SPI1), EWSR1-FLI1, EBF1, ELF5 and RUNX1 (Fig. 3b). The importance of PU.1 binding sites is further supported by data of a ChIP-Seq experiment with CD133+ HPCs which were cultured for 10 days under very similar culture conditions (Fig. 3c)20.

Enrichment of TF-binding motifs.
(a) The sequences within 124 bp around each of the 9,213 overlappingly hypermethylated CpG sites were used for de novo motif analysis. The six most significant motifs were subsequently compared to known TF-binding sites. (b) Alternatively, these sequences were scanned for known motifs and enrichment and p-values (hypergeometric distribution) were calculated in relation to all CpG sites represented on the array. (c) Proportion of PU.1 binding sites within 124, 200, 500 and 1,000 bp around overlappingly hypermethylated CpG sites (black) was enriched compared to all CpG sites of the array (grey; *p < 0.05; ***p < 0.001; Δp < 10−15).
Subsequently, we analyzed gene expression profiles of CD34+ (d0), CD34+ w/o MSC and CD34− w/o MSC. Co-cultured cells were omitted as gene expression is prone to highly expressed genes of contaminating MSCs (in contrast to DNAm profiles which are hardly influenced by the few additional DNA-strands)21. Signal intensity in microarrays was used as an indicator for the gene expression level: as expected, highly expressed genes (the top 10%) were hardly methylated at promoter regions, CGIs and shore-regions (Fig. 4a). Differential DNAm of all CpGs was then plotted against differential gene expression demonstrating that particularly hypermethylation in TSS200 and 1st exon was associated with down-regulated gene expression (p < 10−3; Fig. 4b). In tendency, this association of high DNAm with low gene expression and vice versa became also evident by global analysis of all CpG sites although the correlation was only moderate.

DNAm changes are reflected in differential gene expression.
(a) Gene expression profiles were analyzed by microarray technology (n = 3). For 10% of the genes with highest (highly expressed genes) and 10% with least signal intensity (non-expressed genes) we determined average DNAm levels in different gene regions and in relation to CGIs. (b) Scatter plot analysis depicts CpG sites with differential DNAm (p < 0.05) and differential gene expression (p < 0.05). Higher methylation coincides with less gene expression, particularly in CpG sites located in TSS200 and 1st Exon.
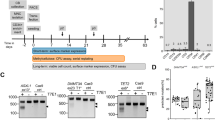
Hypermethylation of DNMT3A might be of specific relevance for induction of further DNAm changes – it was observed at an internal promoter region which did not affect overall gene expression but might influence expression of isoforms (Fig. 5a). In fact, RT-qPCR validated down-regulation of DNMT3A transcripts 2 and 4 upon culture expansion (Fig. 5b). In contrast, DNMT3B revealed neither differential DNAm nor differential gene expression.

DNAm affects splice variants of DNMT3A.
(a) DNAm is presented for CpG sites of DNMT3A and shaded areas depict localisation within the gene. Culture-associated hypermethylation is located at an internal promoter region between transcripts 2 and 4. (b) Quantitative RT-PCR analysis demonstrates that DNMT3A transcripts 2 and 4 are down-regulated upon in vitro expansion of HPCs (n = 5; *p ≤ 0.05; **p ≤ 0.01).
Inhibition of DNMTs modulates expansion of HPCs
To test the influence of demethylating agents on CFU frequency, we used the two cytidine analogues 5-azacytidine (AZA) and zebularine (ZEB), which can be incorporated into the DNA and then covalently bind to catalytic sites of DNMT1, DNMT3A and DNMT3B22. Alternatively, we used epigallocatechin-3-O-gallate (EGCG), which has also been shown to inhibit DNMTs23. Nano-molar doses of DNA-demethylating agents were shown to exert antitumor effects24 and therefore we tested a broad range of concentrations. Overall, proliferation and the percentage of CD34+ cells decreased with higher doses of inhibitors (Supplementary Fig. S5a,b). However, very low concentrations (0.1 μM) of either ZEB (p = 0.0077) or EGCG (p = 0.0009) slightly increased CFU-frequency suggesting enhanced progenitor expansion (Fig. 6).

Treatment with demethylating agents increases CFU-frequency.
HPCs were expanded with various concentrations of 5-azacytidine (AZA), zebularine (ZEB) and epigallocatechin-3-O-gallate (EGCG) and CFU frequency was analyzed after 7 days. Low concentrations of ZEB and EGCG increased CFU-frequency (n = 6; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001). See also Supplementary Fig. S5.
Discussion
In this study, we analyzed DNAm profiles of CD34+ cells and their progeny upon in vitro culture. We demonstrate that HPCs acquire highly reproducible DNAm changes within seven days in vitro. The faster proliferating progeny - which loses CD34 expression - reveals lineage-associated DNAm changes such as hypermethylation in the promoter regions of CD34 and CD133. It was however unexpected, that also cells which maintained CD34+ demonstrated a similar extend of DNA-hypermethylation and this was hardly affected by stromal support. Previous studies have shown that normal hematopoietic differentiation is associated with hypomethylation of lineage specific genes25,26,27,28 and that DNA-methylation may protect from aberrant transcription factor activation29,30. In this regard, the culture-associated hypermethylation observed in this study does not seem to reflect normal hematopoietic differentiation but it rather resembles a roadblock for differentiation towards specific lineages.
It needs to be pointed out, that the CD34+ fraction is relatively heterogeneous and does not resemble pure stem cells. In fact, recent studies indicated that the most primitive HSCs are even CD34−31. In our previous work, we have demonstrated that the culture conditions used in this study maintain NOD/SCID repopulating cells13 – but we did not perform primary or secondary competition transplantation experiments to ultimately address preservation of long-term repopulating HSCs (LT-HSCs). Expression of the myeloid marker CD13 in many freshly isolated and culture expanded CD34+ cells supports the notion that our cell preparations comprise also many more differentiated progenitor cells – although this surface marker may also be expressed on primitive HSCs32. It is conceivable, that culture-associated DNAm changes differ in highly purified LT-HSCs. To this end, it is important to further optimize DNAm profiling technology for picogram DNA amounts obtained from a few thousand cells.
The prevailing DNA-hypermethylation in HPCs during in vitro culture implies that de novo methyltransferases are involved in this process. Conditional ablation of Dnmt3a in mice has been shown to increase the stem cell pool and to impair differentiation33. The relevance of DNMT3A is further supported by frequent mutations in acute myeloid leukemia (AML)34 and myelodysplastic syndromes (MDS)35. We have recently observed that aberrant DNA-hypermethylation frequently occurs within the gene DNMT3A in AML patients thereby mimicking genomic mutations in DNMT3A (Jost et al., in revision). Notably, in this study we describe that the same internal promoter region of DNMT3A is also hypermethylated in HPCs upon in vitro culture. This hypermethylation seems to be associated with down-regulation of the isoforms 2 and 4. Intragenic methylation has previously been shown to regulate alternative promotors36. Transcript 4 does not have methyltransferase activity but its down-regulation may still be functionally relevant if it contributes to regulative complexes. It is also intriguing that DNAm changes were enriched in HOX genes. The HOXA and HOXB clusters are relevant for hematopoietic development and their DNA-hypermethylation has been implicated in various hematopoietic malignancies37. Our analysis revealed that hypermethylated CpG sites are often associated with a HOXA5 binding motif and with binding sites for PU.1 and RUNX1. PU.1 together with GATA1 is involved in hematopoietic cell fate decisions38 and has been shown to interact with DNMT3A/B39. RUNX1 may interact with DNMT140 for site specific DNAm. Simultaneous depletion of RUNX1 and the polycomb group protein PCGF1 allows sustained self-renewal while blocking differentiation of lineage marker-negative cells in vitro41. These findings support the notion that HOXA5, PU.1 and RUNX1 are involved in regulation of DNAm changes during culture of HPCs.
The results of this study point to distinct DNA-hypermethylation of HPCs during in vitro culture. Therefore, it appears to be obvious to test if this process can be blocked by DNAm inhibitors. In fact, low doses of AZA have previously been shown to support expansion of CD34+ cells in vitro42 and combination of AZA with trichostatin A supports maintenance of NOD/SCID mice repopulating cells43. We demonstrate that low doses of ZEB and EGCG facilitate a moderate but significant increase in CFU frequency. Several authors have analyzed the effect of DNMT inhibitors on DNAm profiles in normal HPCs and leukemic cells24,44. For example, Tsai and co-workers have analyzed DNAm profiles of CD34+ cells treated with or without 10 nM Decitabine24. This treatment resulted in an overall decrease of DNAm level with preference for specific genomic regions. However, when we compared DNAm changes in CD34+ cells upon treatment with this DNMT inhibitor and upon culture expansion there was no significant correlation (results not shown). This may be expected, as the mechanism of chromatin modifying agents is different and not site specific. To this end, a better understanding of the functional complexes which govern differential DNAm upon culture of HPCs is required. Direct targeting of these complexes might ultimately block functional changes in culture.
Taken together, in vitro culture of HPCs is associated with loss of differentiation potential in most of the progeny, even though the immunophenotypic characteristics may be maintained. Shared expression of CD34 – and of other surface markers - does not provide any reliable means of saying these cells are functionally similar. We demonstrate that in vitro culture of HPCs is associated with highly reproducible DNA-methylation changes. They are particularly observed in hematopoietic genes and we provide evidence that specific regulatory complexes, such as PU.1, RUNX1, HOXA5 and specific isoforms of DNMT3A, are involved in this process. These epigenetic modifications are reflected on gene expression level and they may interfere with subsequent differentiation. The specific epigenetic modifications may also be relevant for localisation of viral integration sites in the genome of CD34+ cells in gene therapy. Our experimental design does not allow conclusions on DNAm changes in purified HSCs – further research is required to determine whether the small subset of LT-HSCs undergoes epigenetic modifications in vitro such as CD34+ cells. However, the findings of this study indicate that in vitro manipulation of HPCs is associated with specific DNAm changes which seem to be associated with loss of differentiation potential.
Methods
Isolation of HPCs and culture conditions
CD34+ cells were isolated from fresh umbilical cord blood after written consent according to the guidelines specifically approved by the Ethic Committee of RWTH Aachen University (Permit Number: EK187/08) using the CD34 Micro Bead Kit on a MiniMACS system (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany). HPCs were cultured in StemSpan serum free expansion medium (STEMCELL Technologies, Grenoble, France) supplemented with 10 μg/mL heparin (Ratiopharm GmbH, Ulm, Germany), 20 ng/mL thrombopoietin (PeproTech GmbH, Hamburg, Germany), 10 ng/mL stem cell factor (PeproTech), 10 ng/mL fibroblast growth factor 1 (PeproTech). Culture was either performed on tissue culture plastic (TCP) or on a confluent layer of MSCs (passage 3 to 6). Mesenchymal stromal cells were cultured in Dulbecco's Modified Eagle's Medium-Low Glucose (PAA, Pasching, Austria) supplemented with 2 mM l-glutamine (Sigma, St Louis, MO, USA), 100 U/mL penicillin/streptomycin (Lonza, Basel, Switzerland), 10% pooled human platelet lysate45 and 2 U/mL heparin (Roche GmbH, Mannheim, Germany)46. Isolation and characterization of bone marrow derived MSCs was performed as described before13. After seven days, CD34+ and CD34− fractions were again separated as described above. In some experiments, we added the DNMT-inhibitors 5-azacytidine (Sigma-Aldrich), zebularine (Enzo Life Sciences, Farmingdale, NY, USA) and epigallocatechin-3-O-gallate (Enzo Life Sciences) as indicated in Fig. 6.
CFSE staining
Freshly isolated CD34+ cells were labelled with carboxyfluorescein diacetate N-succinimidyl ester (CFSE; Sigma-Aldrich, Steinheim, Germany) to monitor the number of cell divisions. In brief, CD34+ cells were washed with PBS and stained with CFSE at a final concentration of 2.5 μM in PBS with 0.1% fetal calf serum (FCS; PAA, Pasching, Austria) for 10 min at 37°C as described before7,47. To estimate fold-expansion rates of CD34+, we cultured 1,000 CD34+ cells with or without MSCs. After seven days, we analyzed the cell numbers with a CASY cell counter (Schärfe System, Reutlingen, Germany) and multiplied them with the percentage of CD34+ cells.
Flow cytometric analysis
HPCs were stained with propidium iodide (PI), CD34-APC (Becton Dickinson, San Jose, CA, USA [BD], clone 581), CD133/2-PE (Miltenyi Biotec GmbH, clone AC141), CD45-V500 (BD, clone HI30), CD13-PE (BD, clone WM15), CD19-FITC (BD, clone HIB19), CD3-PerCP-Cy5.5 (BD, clone UCHT1), CD56-PE (BD, clone B159) with or without CFSE as indicated in the text. Cells were analyzed using a FACS Canto II (BD) running FACS Diva software (BD).
Colony forming unit assays
10,000 CD34+ cells were cultured for one week as indicated in the text and subsequently re-seeded in different dilutions (1:10 and 1:100 dilution) in methylcellulose (HSC-CFU lite with EPO; Miltenyi Biotec)13. CD34+ (d0) cells were covered with methylcellulose directly after isolation. Granulocyte (CFU-G), macrophage (CFU-M) and erythrocyte colonies (CFU-E) were counted according to manufacturer's instructions.
DNAm profiling
DNAm profiles were analyzed for three corresponding subsets of freshly isolated CD34+ cells (d0), CD34+ w/o MSC, CD34− w/o MSC and CD34+ w/MSC. Genomic DNA was isolated from 106 cells using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) and subsequently bisulfite-converted (EZ DNA Methylation™ Kit; Zymo, Irvine, USA). DNAm profiles were then analyzed with InfiniumHumanMethylation450 BeadChips (Illumina, San Diego, USA) according to the manufacturer's instructions. Hybridization and initial data analysis with the BeadStudio Methylation Module was performed at the DKFZ Gene Core Facility (Heidelberg, Germany). Raw data have been deposited at the public database Gene Expression Omnibus (GEO) under the accession number GSE40799.
For each of the 485 577 CpG sites, DNAm is provided as beta-values ranging from 0 (non-methylated) to 1 (100% methylation). Box plots and histograms were calculated in R. For statistical analysis, we confined 50% of the CpG sites according to variation in DNAm across different samples and calculated adjusted p-values using limma paired t-test in R (P < 0.05; adjusted for multiple testing). Affiliation of CpG sites to gene regions or CGIs was used as described in detail before48 and enrichment was determined by hypergeometric distribution. For graphical presentation of DNAm in specific genes, we compiled all CpG sites related to the corresponding genomic regions. For clarity reasons, the mean of three biological replica is presented although the inter-experimental variation was very small.
Gene Ontology classification of genes with either hypermethylated or hypomethylated CpG sites was performed with GoMiner software (http://discover.nci.nih.gov/gominer/). Enrichment of specific categories was estimated by one-sided Fisher's exact p-value using all CpG sites represented on the array as a reference. Alternatively, we used Gene Set Enrichment Analysis which provided similar results (GSEA; http://www.broadinstitute.org/gsea/index.jsp).
Motif analysis was performed with different programs of the MEME suit (http://meme.nbcr.net). De novo motif discovery was performed with DREME 3, an algorithm for identification of short, core DNA-binding motifs of transcription factors (TFs) within 124 bp around hypermethylated CpG sites. The most significant motifs were subsequently used for the TOMTOM motif comparison tool to identify potential TFs which bind to these motifs. Furthermore, we scanned for the occurrence of given motifs using the FIMO tool 4 within 124 bp around hypermethylated CpG sites (alternatively, we have extended these regions to 2 kb, but the high number of TF-binding sites made these results less specific). Enrichment was estimated in relation to all CpG sites represented on the array (hypergeometric distribution). Binding sites for PU.1, predicted by ChIP-Seq on cultured HPCs, were obtained from Novershtern et al20. Enrichment of PU.1 binding sites close to hypermethylated CpG sites was calculated by Fisher's exact test. We tested co-location window-values of 124, 200, 500 and 1,000 bp around hypermethylated CpG sites and used all CpGs in the array as background.
DNAm analysis by bisulfite pyrosequencing
Genomic DNA was isolated and bisulfite-converted as described above. Specific regions within CD34 and DNMT3A genes were then amplified by PCR and 20 μl of the product was immobilized to 5 μl Streptavidin beads followed by annealing to 0.8 μl sequencing primer (20 μM) for 2 min at 80°C. Primer information is provided in Supplementary table S1. Pyrosequencing was then performed on a PyroMark Q96 ID System and results were analyzed with the PyroMark Q CpG software (Qiagen).
Gene expression analysis
Gene expression profiles were analyzed in three additional CB samples for CD34+ (d0), CD34+ w/o MSC and CD34− w/o MSC. RNA was isolated from 106 cells using the miRNeasy Mini Kit (Qiagen). Quality control and measurement of RNA concentration was done with a NanoDrop Spectrophotometer (Thermo Scientific, Wilmington, USA) and the Agilent 2100 Bioanalyzer (Agilent Technologies). Gene expression profiles were determined on GeneChip Human Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's instructions. Raw data were normalized by RMA (Affymetrix Power Tools) and matched to DNAm data by gene ID. Data are accessible at GEO under GSE40669.
RT-qPCR analysis of DNMT3A transcripts
Expression of DNMT3A transcripts was analyzed by real-time quantitative PCR (RT-qPCR) using the StepOneTM Instrument (Applied Biosystems, Applera Deutschland GmbH, Darmstadt, Germany). Total RNA (250 ng) was reverse-transcribed using the high capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA was diluted 1:2 with nuclease-free water and 2 μl were amplified using Power SYBR Green PCR Master Mix (Applied Biosystems). Gene expression was normalized to GAPDH. The following primers (Metabion, Martinsried, Germany) were used for amplification of DNMT3A transcripts: Transcripts 1 and 3: Forward: 5′-ACCCTGCCTGAAGCCTCAAG-3′, Reverse: 5′-AAGGTGAGCCTCGGCATGG-3′; Transcript 2: Forward: 5′-GTGGATCGTAGCCTGAAAG-3′, Reverse: 5′-TGGTGGCATTCTTGTCC-3′; Transcript 4: Forward: 5′-AAGCGGGTGAGTCCTCAGC-3′, Reverse: 5′-CATATGCGCAGGCTGCATCC-3′.
Statistics
All results are indicated with standard error of the mean (SEM). To estimate the probability of differences, we have adopted the two-sided Student's T-test. Probability value of p < 0.05 denoted statistical significance.
References
Ballen, K. K., Gluckman, E. & Broxmeyer, H. E. Umbilical cord blood transplantation: the first 25 years and beyond. Blood 122, 491–498 (2013).
Naldini, L. Ex vivo gene transfer and correction for cell-based therapies. Nat. Rev. Genet. 12, 301–315 (2011).
Zhang, C. C., Kaba, M., Iizuka, S., Huynh, H. & Lodish, H. F. Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood 111, 3415–3423 (2008).
Boitano, A. E. et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 329, 1345–1348 (2010).
Delaney, C. et al. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 16, 232–236 (2010).
Robinson, S. N. et al. Superior ex vivo cord blood expansion following co-culture with bone marrow-derived mesenchymal stem cells. Bone Marrow Transplant. 37, 359–366 (2006).
Walenda, T. et al. Co-Culture with Mesenchymal Stromal Cells Increases Proliferation and Maintenance of Hematopoietic Progenitor Cells. J. Cell Mol. Med. 14, 337–350 (2010).
Jing, D. et al. Hematopoietic stem cells in co-culture with mesenchymal stromal cells--modeling the niche compartments in vitro. Haematologica 95, 542–550 (2010).
Okano, M., Bell, D. W., Haber, D. A. & Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Yamagata, Y. et al. Lentiviral transduction of CD34(+) cells induces genome-wide epigenetic modifications. PLoS One 7, e48943 (2012).
Bibikova, M. et al. High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295 (2011).
Walenda, T. et al. Synergistic effects of growth factors and mesenchymal stromal cells for expansion of hematopoietic stem and progenitor cells. Experimental Hematology 39, 617–628 (2011).
Koch, C. M. et al. Monitoring of Cellular Senescence by DNA-Methylation at Specific CpG sites. Aging Cell 11, 366–369 (2012).
Koch, C. et al. Pluripotent Stem Cells Escape From Senescence-Associated DNA Methylation Changes. Genome Res. 23, 248–259 (2013).
Stier, S., Cheng, T., Dombkowski, D., Carlesso, N. & Scadden, D. T. Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 99, 2369–2378 (2002).
Sauvageau, G. et al. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 9, 1753–1765 (1995).
Schiedlmeier, B. et al. HOXB4's road map to stem cell expansion. Proc. Natl. Acad. Sci. U. S. A 104, 16952–16957 (2007).
Irizarry, R. A. et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 41, 178–186 (2009).
Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309 (2011).
Wagner, W. et al. Hematopoietic progenitor cells and cellular microenvironment: behavioral and molecular changes upon interaction. Stem Cells 23, 1180–1191 (2005).
Champion, C. et al. Mechanistic insights on the inhibition of c5 DNA methyltransferases by zebularine. PLoS ONE 5, e12388 (2010).
Fang, M. Z. et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 63, 7563–7570 (2003).
Tsai, H. C. et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 21, 430–446 (2012).
Yu, Y. et al. High resolution methylome analysis reveals widespread functional hypomethylation during adult human erythropoiesis. J Biol Chem. 288, 8805–8814 (2013).
Broske, A. M. et al. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 41, 1207–1215 (2009).
Hogart, A. et al. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal overrepresentation of ETS transcription factor binding sites. Genome Res. 22, 1407–1418 (2012).
Calvanese, V. et al. A promoter DNA demethylation landscape of human hematopoietic differentiation. Nucleic Acids Res. 40, 116–131 (2012).
Bock, C. et al. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 47, 633–647 (2012).
Ji, H. et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342 (2010).
Anjos-Afonso, F. et al. CD34(−) cells at the apex of the human hematopoietic stem cell hierarchy have distinctive cellular and molecular signatures. Cell Stem Cell 13, 161–174 (2013).
Taussig, D. C. et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood 106, 4086–4092 (2005).
Challen, G. A. et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 44, 23–31 (2012).
Ley, T. J. et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 (2010).
Walter, M. J. et al. Recurrent DNMT3A mutations in patients with myelodysplastic syndromes. Leukemia 25, 1153–1158 (2011).
Maunakea, A. K. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 (2010).
Strathdee, G. et al. Inactivation of HOXA genes by hypermethylation in myeloid and lymphoid malignancy is frequent and associated with poor prognosis. Clin. Cancer Res. 13, 5048–5055 (2007).
Wolff, L. & Humeniuk, R. Erythroid versus Myeloid Lineage Commitment: Regulating the Master Regulators. Stem Cells 31, 1237–1244 (2013).
Suzuki, M. et al. Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 25, 2477–2488 (2006).
Liu, S. et al. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res. 65, 1277–1284 (2005).
Ross, K. et al. Polycomb group ring finger 1 cooperates with Runx1 in regulating differentiation and self-renewal of hematopoietic cells. Blood 119, 4152–4161 (2012).
Suzuki, M. et al. 5-Azacytidine supports the long-term repopulating activity of cord blood CD34(+) cells. Am. J. Hematol. 77, 313–315 (2004).
Milhem, M. et al. Modification of hematopoietic stem cell fate by 5aza 2'deoxycytidine and trichostatin A. Blood 103, 4102–4110 (2004).
Stresemann, C., Bokelmann, I., Mahlknecht, U. & Lyko, F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol. Cancer Ther. 7, 2998–3005 (2008).
Schallmoser, K. et al. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion 47, 1436–1446 (2007).
Hemeda, H., Kalz, J., Walenda, G., Lohmann, M. & Wagner, W. Heparin concentration is critical for cell culture with human platelet lysate. Cytotherapy 15, 1174–1181 (2013).
Walenda, T. et al. Serum after autologous transplantation stimulates self-renewal and proliferation of human hematopoietic progenitor cells. PLoS ONE 6, e18012 (2011).
Sandoval, J. et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 6, 692–702 (2011).
Acknowledgements
We are grateful to Matthias Schick (gene core facility at the German Cancer Research Center, Heidelberg, Germany) for array hybridization and initial data analysis. This work was supported by the Stem Cell Network of the state North Rhine Westphalia, by the German Research Foundation (DFG; WA 1706/2-1) and by the German Ministry of Education and Research (BMBF; CB-HERMES). The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Contributions
C.W., T.W., Q.L., B.D., I.C., M.Z. and W.W. designed research, analyzed and interpreted data; C.W., T.W. and Q.L. performed experiments; M.W., B.D. and M.Z. contributed vital reagents; C.W. and W.W. wrote the manuscript and all authors provided input to the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Weidner, C., Walenda, T., Lin, Q. et al. Hematopoietic Stem and Progenitor Cells Acquire Distinct DNA-Hypermethylation During in vitro Culture. Sci Rep 3, 3372 (2013). https://doi.org/10.1038/srep03372
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03372
This article is cited by
-
Reconstruction of bone marrow microenvironment for expansion of hematopoietic stem cells by a histone deacetylase inhibitor
Cytotechnology (2023)
-
Hematopoietic differentiation persists in human iPSCs defective in de novo DNA methylation
BMC Biology (2022)
-
DNA methylation dynamic of bone marrow hematopoietic stem cells after allogeneic transplantation
Stem Cell Research & Therapy (2019)
-
Therapeutic potential of menstrual blood-derived endometrial stem cells in cardiac diseases
Cellular and Molecular Life Sciences (2019)
-
Epigenetic aging of human hematopoietic cells is not accelerated upon transplantation into mice
Clinical Epigenetics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
