
Abstract
CRISPR/Cas mediated genome editing has been successfully demonstrated in mammalian cells and further applications for generating mutant mice were reported by injecting humanized Cas9 (hCas) mRNA and single guide RNA into fertilized eggs. Here we inject the circular plasmids expressing hCas9 and sgRNA into mouse zygotes and obtained mutant mice within a month. When we targeted the Cetn1 locus, 58.8% (10/17) of the pups carried the mutations and six of them were homozygously mutated. Co-injection of the plasmids targeting different loci resulted in the successful removal of the flanked region in two out of three mutant pups. The efficient mutagenesis was also observed at the Prm1 locus. Among the 46 offspring carrying CRISPR/Cas plasmid mediated mutations, only two of them carried the hCas9 transgene. The pronuclear injection of circular plasmid expressing hCas9/sgRNA complex is a rapid, simple and reproducible method for targeted mutagenesis.
Similar content being viewed by others


Introduction
Gene knockout animals are a robust tool for elucidating the roles of numerous genes in development, growth, adult physiology and disease1. For designed mutagenesis, the drug resistant gene has been traditionally introduced into the genome through homologous recombination in embryonic stem (ES) cells, chimeric mice production and germ-line transmission by mating experiments2. Although it is a widely used approach, it is laborious, costly and time consuming. Moreover, only well trained researchers are able to accomplish all experimental procedures.
The emergence of zinc-finger nucleases (ZFN) and/or transcription activator-like effector nucleases (TALEN) have opened the window for the next generation of targeted mutagenesis3. These enzymes are artificially generated by fusing FokI endonucleases with DNA recognition motifs. The enzymes recognize target DNA by peptide-DNA affinity and fused FokI nucleases generate double strand breaks (DSB), subsequently error-prone non-homologous end joining (NHEJ) results in small indels3. Moreover, if reference ssDNA or dsDNA exists, homology dependent repair (HDR) or high-fidelity homologous recombination (HR) introduces designed mutations into the targeted locus4. Since the DSB mediated mutation is efficient, one-step generation of gene targeted mice and rats have been reported by injecting the mRNA coding these enzymes into zygotes5,6. However, the difficulty in the design and preparation of these enzymes hampered the spreading of the technique.
Recently, the type II CRISPR (Clustered regulatry interspaced short palindromic repeat)/Cas (CRISPR associated) system has been demonstrated to cause DSB in mammalian cells7,8. The CRISPR/Cas system was originally found in bacteria and archaea and has turned out to be an RNA-based adaptive immune system to destroy invading plasmids, phages and viruses9,10,11. The nucleoprotein complex consisting of CRISPR coded RNAs (crRNAs), trans-activating crRNAs (tracrRNA) and Cas proteins, recognize foreign DNA by the crRNA sequences and degrade it by endonuclease activity12. It is noteworthy that the combination of the humanized Cas9 (hCas9) protein with synthetic single-guide RNA (sgRNA) generated by fusing crRNA and tracrRNA could reconstitute RNA-based nucleases and cause DSB in mammalian cells7,8. The indels caused by error-prone NHEJ leads to targeted gene mutation.
In the present report, we have developed a simple validation system for the gene targeted DSB by observing green fluorescence reconstituted by HDR of an EGFP expression cassette (Fig. 1a). Different from ZFNs and TALENs, gene targeting ribonuclear complexes can easily be designed and prepared by changing sgRNA sequence. Thus we used the single plasmid, pX3307, containing a hCas9 expression cassette with a gene targeting sgRNA expression cassette. After the validation in vitro, we injected the plasmid into fertilized mouse eggs in it's circular form to decrease the chance of integration into the genome. Finally, gene targeting efficiency and transgenicity were examined as well as off-target cleavages. Whereas Wang et al13., demonstrated one-step generation of mice carrying mutations by injecting hCas9 mRNA with sgRNA into zygotes, our method can skip the mRNA and sgRNA synthesis and provide simple and reproducible method for targeted mutagenesis.

Scheme for CRISPR/Cas mediated gene manipulation.
(a) pCAG-EGxxFP plasmid contains 5′ and 3′ EGFP fragments that shares 482 bp under ubiquitous CAG promoter. The ~500 bp genomic fragment containing the sgRNA target sequence was placed between EGFP fragments of pCAG-EGxxFP plasmid. The resulting target plasmid was cotransfected with pX330 plasmids expressing sgRNA and hCas9 into HEK293T cells. When the target sequence was digested by sgRNA guided CAS9 endonuclease, the homology dependent repair (HR; homologous recombination or SSA: single strand annealing) took place and reconstituted the EGFP expression cassette. MCS; multi cloning sites. (b) The plasmids used in the study. pCAG-EGxxFP contains multicloning sites (BamHI, NheI, PstI, SalI, EcoRI and EcoRV). pX330 and pT7-sgRNA plasmids contains BbsI sites that enables directional cloning of sgRNA oligos7. (c) The efficiency of DSB mediated homology dependent repair was validated by observing EGFP fluorescence 48 hrs after the transfection (top; pX330 without sgRNA, bottom; pX330 with Cetn1/sgRNA1). (d) To generate gene disrupted mice, fertilized eggs were injected with RNAs coding hCas9 and sgRNA into cytoplasm or pX330 plasmid into pronuclei.
Results
Preparation of the CRISPR/Cas plasmids for genome engineering
Activity of gene-targeted endonucleases have been traditionally validated by Cel-I nuclease digestion of PCR amplified targeted region and/or the single strand annealing (SSA) assay that reconstitutes reporter gene expression14. Here we prepared the pCAG-EGxxFP plasmid containing 5′ and 3′ EGFP fragments that share 482 bp under ubiquitous CAG promoter15 (Fig. 1b). An approximately 500 bp region of the target genome was inserted between the EGFP fragments and used as a target plasmid. For expressing hCas9 and sgRNA, pX330 plasmid prepared by Dr. Feng Zhang was used7. To validate which sgRNA sequence works, we cotransfected the pCAG-EGxxFP-target and pX330-sgRNA plasmids into HEK293T cells and the reconstituted EGFP fluorescence was observed 48 hrs after transfection (Fig. 1c). With effective sgRNA sequences, more than 30% of the transfected cells became fluorescent. The oligos and primers as we used are listed in Table S1.
One-step generation of gene mutant mice by microinjection into the zygotes
Following validation of sgRNA containing pX330 plasmids in human 293T cells, we used two approaches to generate gene targeted mutant mice by zygote injection (Fig 1d). First, we constructed plasmids in which hCas9 and sgRNA is placed under T7 promoter (Fig. 1b). In vitro transcribed hCas9 mRNA was 5′-capped, 3′-polyadenylated and then injected with sgRNA. Secondly, to minimize the steps and efforts, we directly injected pX330-sgRNA plasmids into pronuclei of fertilized eggs. The pups developed from these eggs were genotyped by PCR and subsequent sequence analysis.
When we examined Centrin 1 (Cetn1) and Protamine 1 (Prm1) loci, microinjection of hCas9 mRNA along with sgRNA efficiently caused indels at the targeted locus (Fig. 2, Table 1). Consistent with the recent report13, the mutation frequency appeared to depend on RNA concentration and all four pups developed from eggs injected with hCas9 mRNA and sgRNA at 40 and 10 ng/ul, respectively, carried the mutation at the Prm1 loci.

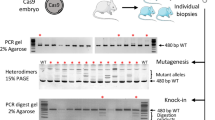
CRISPR/Cas mediated Cetn1 and Prm1 mutations in mice.
(a) Cetn1 mutations observed in founder mice. The small indels were identified at sgRNA targeted locus (bold; sgRNA recognition site, red; PAM sequence, slash; predicted cleavage site and arrow; sgRNA direction). Some mutations were frequently observed in independent founder mice. The numbers of mutants obtained were indicated in parenthesis (left; with plasmid injection, right; with RNA injection). (b) Representative Cetn1 genomic sequences from founder mice (top; wild type, middle; heterozygous 8 bp deletion, homozygous 8 bp deletion). (c and d) Prm1 mutations observed in founder mice.
When the pX330 plasmids were injected at five ng/ul, 58.8% (10/17) of pups carried the targeted mutation at the Cetn1 locus (Table 2). It is noteworthy that we could not detect any wild-type signals in six among the 10 mutants, suggesting these were homozygously mutated at the targeted locus (Fig. 2b). Prior to the further analysis, all the tail tip DNA samples collected from the mutant mice were subjected to PCR analysis. Among the 46 mutants obtained with nine pX330 constructs (Cetn1, Prm1 and 7 other targets), only two pups carried the hCas9 transgene.
CRISPR/Cas mediated deletion between two loci
To examine whether the region flanked by two target loci can be removed, we coinjected the two pX330 plasmids targeting separate regions 381 bp apart in the Cetn1 gene (predicted cleavage sites). Among the three mutants obtained, two carried the deletion that removed the region flanked by the sites targeted by two sgRNAs, sgRNA1 and sgRNA4 (Table 3). Another mutant carried the single mutation at sgRNA1, which reflected the rate of DSB observed in the HEK293T EGFP assay, i.e. sgRNA1 demonstrated a better EGFP assay performance (Table 3).
Off-target analysis in CRISPR/Cas mediated mutants
Although the single nucleotide exchange within the seed 10 bases of sgRNA severely diminished its target specificity7, the risk of off-target mutation remains in Cas9/sgRNA mediated mutant animals. In the present study, we searched the off-target sites that exactly matches 12–13 bases at the 3′ end and the NGG (N can be A, G, C, or T) with free software, Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) against whole mouse genomic sequence (mm9). There were seven and four off target candidates were found in Cetn1/sgRNA1 and Prm1/sgRNA1, respectively. We amplified and sequenced the ~1 kbp regions that contain the potential off-target sites. A total of 144 off-target sites were analyzed (seven off-target sites for 16 Cetn1 mutants and four off-target sites for eight Prm1 mutants) and a single off-target mutation was observed in one of the Cetn1 mutant mice (Table S2).
Male sterility in Cetn1 and Prm1 mutants
Taking advantage of the homozygous mutant mice generated by pX330 injection, we analyzed the Cetn1 homozougous mutant mice. As we expected from the male germ cell specific expression pattern, male Cetn1 mutant (em4/em4; eight bp deletion, d8) mice were infertile (Fig. 3a). Although gross morphology of testis looked normal in the mutant mice, microscopic analysis clarified the impaired ciliogenesis at the spermatid stage and thus malformed spermatozoa that were unable to fertilize eggs were generated (Fig. 3b and Fig. S1). When we analyzed Prm1 mutant mice (wt/em3; five bp deletion, d5), male infertility with sperm malformation and defective sperm motility was observed in mutant founder males, indicating haploinsufficiency of Prm1 gene in mice (Fig. 3c and 3d). These results were consistent with the reports with conventionally generated Cetn1 and Prm1 knockout mice, respectively16,17.

Male infertility found in Cetn1 and Prm1 deficient mice.
(a) Average litter size obtained from Cetn1 mutant males. Each male was mated with two B6D2F1 females for two months and numbers of pups were counted at birth. (b) Sperm collected from epidydimis of 12 weeks old males were photographed under a phase contrast microscopy. Impaired ciliogenesis was observed in Cetn1em4/em4 mice. Scale bar; 20 um. (c) Sperm motility observed with Prm1 mutant males. The eggs were collected from superovulated B6D2F1 females mated with 12 weeks old males. The sperm motility was anlayzed by Ceros system at 10 min and 120 min of incubation. (d) Sperm collected from epidydimis of 12 weeks old males were photographed under a differential interference contrast microscopy. Heads narrowed and reduced in curvature at the tip were observed in most Prm1+/em3 sperm. Scale bar; 20 um.
Discussion
Development of chimeric nucleases, ZFNs and TALENs, opened the next-generation of mammalian genome engineering by inducing a targeted DNA double strand breaks that stimulate error-prone NHEJ or HDR18,19. However, even after several methods such as Golden-gate assembly20 have been developed to decrease the steps, difficulties in preparing the plasmid coding multiple fingers in designed array hampered the process, preventing its widespread use. Meanwhile an emergence of the CRISPR/Cas system made this approach more simple, easy and cost-effective7,8. It depends on an RNA guided target DNA recognition system and the CRISPR/Cas mediated mutagenesis thus far demonstarated in mice has been achieved by injecting hCas9 mRNA with sgRNA into the zygote13,21.
In the present study, we bypassed the RNA preparation that requires careful handling and storage and injected the pX330 plasmid that expresses sgRNA and hCas9 under a U6 and chicken beta actin promoter, respectively7. The efficiency is not able to be directly compared due to the differences in toxicity, stability and expression efficiency, etc between mRNA and plasmids. However, the targeted mutant animals were competently obtained after the pX330 plasmid injection (Table 2). In addition, by using circular plasmid, we minimized the risk of undesired transgene integration into host genome22. Although 4.3% (2/46) of mutant pups carried the hCas9 transgene, the transgenicity is lower than our average transgenic efficiency with linearised DNA, 33.4 ± 23.0% (173/684, N = 26 constructs). Thus we conclude that the direct injection of pX330 into the pronucleus is a simple, easy and fast approach to generate targeted gene knockout mice.
More significantly, we found six out of 10 mutants carried biallelic mutations at the Cetn1 locus. Thus mice carrying homozygous mutation can be generated within a month (one week for pX330 construction and three weeks for founder production). Since conventional gene targeting strateties requires at least eight months to obtain homozygous mutant (one month for ES cell screening, one month for chimera production, two months for chimera maturation, one month for germ line transmission, two months for heterozygote maturation, one month for homozygous fetal development), it very much accelerates the mutant mice generation phase. Not only the laborious work and the expenses, more importantly animal lives are also saved. It should be noted that all six homozygous mutants carried the exactly the same mutations in both alleles (one for d1/d1, two for d8/d8, two for d15/d15 and one for d135/d135). The genomic DNA hit by a second DSB might refer the firstly mutated DNA to repair. And/or, as suggested in Wang's paper13, same micro homology dependent repair could occur simultaneously within a zygote. These are consistent with our data that the some mutations are frequently generated independently (Fig. 2a and 2c).
We also showed that multiple DSBs at a different locus occurs in a zygote. If the target sites are close, the flanked regions were efficiently removed between target sites (Table 3). Since it is very time-consuming to target two close loci with the traditional ES cell mediated approach (two rounds of homologous recombination with different drug selection cassettes), CRISPR/Cas system provides a powerful tool for mutating the clustered genes. The targeting of multi-loci over different chromosomes was reported13. Interestingly, single mutations with small indels were more frequently observed in the site which was targeted more efficiently in vitro (Table 3). This result implicates that the validation in 293T human cell line is useful prior to murine zygote injection.
As seen in ZFNs and TALENs, off-target cleavages have been questioned in the CRISPR/Cas system. In the present study, we found only one off-targeted mutation in a total of 144 sites examined. Although it is difficult to compare the off-target risk among these artificial nucleases, one could argue that ZFNs and TALENs are safer because they require two adjacent recognition sites while CRISPR/Cas requires only one target site. On the other hand, whereas ZFNs and TALENs depend on peptide-nucleotide recognition, CRISPR/Cas system uses RNA-DNA affinity in which the affinity is high and easily calculated. Recently, the risk of off-target was claimed with in vitro studies23,24. Different from the continuous and excess expression in cultured cell lines, transient expression by pronuclear injection could decrease this risk. The amount of expression is also easily regulated by changing the concentratio of plasmid to be injected into the zygote. Since each sgRNA has different off-targets, analysis of an independent line established with different sgRNAs can minimize the risk of off-target cleavage effects on the phenotype.
Finally we could observe the effects of Cetn1 and Prm1 deletion in spermatogenesis. As reported previously16, Prm1 mutant mice showed a haploinsuficiency phenotype, chromatin mis-folding and subsequent malformation of the sperm head. As for Cetn1, the homozygous mutant (em4/em4) showed male infertility with defective sperm tail elongation, thus generating sperm with impaired motility. Similar phenotypes were recently reported during our manuscript preparation17. As we have shown here, the phenotypes observed in the CRISPR/Cas mediated mutants were the same as in knockcout mice conventionally generated through ES cells. Here we conclude the pronuclear injection of circular plasmid expressing Cas9/gRNA complex is a rapid, simple and reproducible method for the targeted mutagenesis in mice and can propel the in vivo gene function study furhter than ever before. Moreover, athough the large scale knockout mice projects are ongoing, thousands of genes (e.g. sex chromosome linked genes) remained to be targeted because of the difficulties in vector constrution and gene targeting in ES cells. We believe that our simple method provides the solution to complement the current knockout project that is beneficial for the biological/biomedical researchers worldwide.
Methods
Animals
All animal experiments were approved by the Animal Care and Use committee of the Research Institute for Microbial Diseases, Osaka University.
Plasmid and mRNA preparation
To construct pCAG-EGxxFP validation plasmid, N-terminal and C-terminal EGFP coding regions were PCR amplified and placed under ubiquitous CAG promoter with multicloning sites, BamHI, NheI, PstI, SalI, EcoRI and EcoRV. The ~500 bp genomic fragments containing sgRNA target sequence were PCR amplified and placed between the EGFP fragments. The plasmids expressing hCas9 and sgRNA were prepared by ligating oligos into BbsI site of pX330 (http://www.addgene.org/42230/)7. As for RNA preparation, sgRNA and hCas9 sequences were removed from pX330 and placed under T7 promoter in pUC19 plasmids and named as pT7-sgRNA and pT7-hCas9, respectively. The resulting plasmids were subjected to RNA synthesis with mMESSAGE mMACHINE T7 kit (Ambion, Austin, TX) with manufacturer's protocol. The hCas9 mRNA was polyadenlylated with polyA tailing kit (Ambion, Austin, TX) prior to purification with RNeasy kit (QIAGEN, Chatsworth, CA). The sgRNAs were purified by phenol-chloroform-isoamylalchol extraction and isopropanol precipitation followed by spin column chromatography with MicroSpin G-25 (GE Healthcare, Milwaukee, WI).
HEK293T cells transfection
Five handred ng of pCAG-EGxxFP-target was mixed with 500 ng of pX330 with/without sgRNA sequences and then introduced into 4 × 105 HEK293T cells/well in six well plate by the conventional calcium phosphate transfection method. The EGFP fluorescence was observed under fluorescence microscope at 48 hrs after transfection.
Pronuclear injection
B6D2F1 female mice were superovulated and mated with B6D2F1 males and fertilized eggs were collected from the oviduct. The pronuclear stage eggs were injected with pX330 plasmids, hCas9 mRNA and sgRNAs at indicated concentrations. The eggs were cultivated in kSOM overnight then transferred into the oviducts of pseudopregnant ICR females.
Off target analysis
Potential off-target sites were found using a free software, Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) with rules outlined previously8,13. Twelve to Thirteen bases preceding the PAM sequence with AGG, GGG, CGG and TGG were aligned with mouse genome (mm9). The ~1 kb genomic fragments containing the off-target in the center were PCR amplified and sequenced.
Male fertility test
Sexually matured Cetn1 mutant male mice were caged with two month-old B6D2F1 female mice for two months and the number of pups was counted at the day of birth. As for Prm1 mutant males, superovulated B6D2F1 females were mated with Prm1 mutant males then the eggs were collected from copulated females. The fertilization event was confirmed by pronuclear formation at 20 hrs after hCG injection.
Analysis of sperm motility
Epididymal sperm were collected and incubated in a drop of TYH medium25 covered with liquid paraffin (Nacalai tesque, Kyoto, Japan). The suspension was then diluted 1:100 in TYH medium prior to motility measurement. Sperm motility was measured using the CEROS sperm analysis system (software version 12.3; Hamilton Thorne Biosciences, Beverly, MA). Analyasis setting described previously26 was used.
References
Guan, C., Ye, C., Yang, X. & Gao, J. A review of current large-scale mouse knockout efforts. Genesis 48, 73–85 (2010).
Skarnes, W. C. et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 (2011).
Gaj, T., Gersbach, C. A. & Barbas, C. F., 3rd ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31, 397–405 (2013).
Menke, D. B. Engineering subtle targeted mutations into the mouse genome. Genesis 51, 605–618 (2013).
Mashimo, T. et al. Efficient gene targeting by TAL effector nucleases coinjected with exonucleases in zygotes. Sci Rep 3, 1253 (2013).
Sung, Y. H. et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31, 23–24 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Garneau, J. E. et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 (2010).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109, E2579–2586 (2012).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Makarova, K. S. et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9, 467–477 (2011).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013).
Oleykowski, C. A., Bronson Mullins, C. R., Godwin, A. K. & Yeung, A. T. Mutation detection using a novel plant endonuclease. Nucleic Acids Res 26, 4597–4602 (1998).
Okabe, M., Ikawa, M., Kominami, K., Nakanishi, T. & Nishimune, Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett 407, 313–319 (1997).
Cho, C. et al. Haploinsufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet 28, 82–86 (2001).
Avasthi, P. et al. Germline deletion of Cetn1 causes infertility in male mice. J Cell Sci 126, 3204–3213 (2013).
Miller, J. C. et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25, 778–785 (2007).
Miller, J. C. et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29, 143–148 (2011).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39, e82 (2011).
Li, D. et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31, 681–683 (2013).
Araki, K., Araki, M., Miyazaki, J. & Vassalli, P. Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A 92, 160–164 (1995).
Pattanayak, V. et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31, 839–843 (2013).
Fu, Y. et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31, 822–826 (2013).
Toyoda, Y., Yokoyama, M. & Hoshi, T. Studies on the fertilization of mouse eggs in vitro. Jpn J Anim Reprod 16, 147–151 (1971).
Goodson, S. G., Zhang, Z., Tsuruta, J. K., Wang, W. & O'Brien, D. A. Classification of mouse sperm motility patterns using an automated multiclass support vector machines model. Biol Reprod 84, 1207–1215 (2011).
Acknowledgements
We thank Dr. F. Zhang for pX330 plasmid, Y. Esaki, S. Nishioka and H. Kato for technical assistance in generating mutant mice, BSc (Honors). S.A.M. Young for critical reading and Dr. M. Okabe for discussion and encouragement. This work was supported in part by the MEXT of Japan.
Author information
Authors and Affiliations
Contributions
D.M., Y.F. and M.I. performed most experiments, assisted by Y.S., H.M. and A.I. D.M., Y.F. and Y.S. generated mutant mice. D.M., Y.F. and M.I. analyzed the data. M.I. wrote the manuscript and all authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Mashiko, D., Fujihara, Y., Satouh, Y. et al. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 3, 3355 (2013). https://doi.org/10.1038/srep03355
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03355
This article is cited by
-
Synthetic circuits based on split Cas9 to detect cellular events
Scientific Reports (2023)
-
Identification of a novel type of focal adhesion remodelling via FAK/FRNK replacement, and its contribution to cancer progression
Cell Death & Disease (2023)
-
An Igh distal enhancer modulates antigen receptor diversity by determining locus conformation
Nature Communications (2023)
-
VGLL3 is a mechanosensitive protein that promotes cardiac fibrosis through liquid–liquid phase separation
Nature Communications (2023)
-
The engineered single guide RNA structure as a biomarker for gene-editing reagent exposure
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
