
Abstract
Diabetes is caused by the loss or dysfunction of insulin-secreting β-cells in the pancreas. β-cells reduce their mass and lose insulin-producing ability in vitro, likely due to insufficient cell-cell and cell-extracellular matrix (ECM) interactions as β-cells lose their native microenvironment. Herein, we built an ex-vivo cell microenvironment by culturing primary β-cells in direct contact with ‘synthetic neighbors', cell-sized soft polymer microbeads that were modified with cell-cell signaling factors as well as components from pancreatic-tissue-specific ECMs. This biomimetic 3D microenvironment was able to promote native cell-cell and cell-ECM interactions. We obtained sustained maintenance of β-cell function in vitro enhanced cell viability from the few days usually observed in 2D culture to periods exceeding three weeks, with enhanced β-cell stability and insulin production. Our approach can be extended to create a general 3D culture platform for other cell types.
Similar content being viewed by others


Introduction
Cells exist in vivo within the complex microenvironment that makes up their native tissue, from which they receive a continuous supply of nutrients and to which they release waste; they undergo tissue-specific interactions and signaling with extracellular matrix (ECM) components and communication with neighboring cells1,2,3. Current in vitro cell culture removes cells from their native tissue context and places them on a 2D surface in culture flasks, which can disrupt these interactions and induce changes in gene expression and cellular phenotype1. In order to address these limitations, researchers have investigated different approaches to 3D cell culture using biocompatible materials for microencapsulation, microparticles or cell-laden hydrogels modified with ECM proteins4,5,6,7 with improved function8,9,10; however, even the most advanced in vitro 3D culture approaches lack important features needed to reconstitute their in vivo counterparts.
The β-cells in the pancreatic islets regulate their secretion of insulin in response to glucose levels in the blood to maintain glucose homeostasis in the body. In the islets, β-cells occupy over 60% percent of the total volume11. Direct contact between cells and cell–cell interactions are important for many cellular activities to maintain survival and function of β-cells12,13,14, including intracellular signaling. Recently, Konstantinova et al. showed that β-cells communicate via EphA receptors and EphrinA ligands15. Based on this finding, the Anseth group achieved better survival and insulin secretion of β-cells over ten days by encapsulating the cells into EphA–EphrinA and cell-adhesive peptide (RGD) functionalized poly(ethylene glycol) (PEG) hydrogels16. However, encapsulation of cells within hydrogels may lead to cell death due to diffusional limitations in oxygen supply and nutrients17. Furthermore, exposure of cells to the harsh chemical (i.e. pH change, or high ion concentration) or physical (UV irradiation) environments used during many encapsulation processes is cytotoxic and may affect cellular activity18. A challenge is that traditional bulk homogeneous hydrogel constructs cannot provide a truly 3D environment that effectively replaces cell-cell interactions.
Herein, we propose a new strategy for engineering an ex vivo 3D microenvironment for studying the stability and function of pancreatic β-cells for which microgels are designed as “synthetic neighbors” capable of presenting ligand and replicating aspects of the cell-cell interactions between beta cells in a crowded cell environment such as the pancreas. Our goal is to build an artificial 3D home for β-cells that can thus recapitulate the native tissue conditions in pancreatic islets. Specifically, as shown in Figure 1, β-cells are cultured in direct contact with soft microbeads that are similar in size and mechanical property to cells. These microbeads are made from crosslinked poly(ethylene glycol)-co-poly-L-lysine (PEG-co-PLL) hydrogels modified with the cell surface receptor and its membrane-bound ligand pair, EphA/EphrinA and coated with pancreatic tissue specific ECM components derived from rat pancreatic decellularized matrix. In contrast to direct cell encapsulation in PEG gels, we can place β-cells together with microbeads to create a 3D culture condition in which the β-cells are surrounded by synthetic neighbor cells that present the key ligands and receptors needed for cell-cell communication as well as appropriate matrix. Furthermore, because cells are not fixed in the hydrogel networks, they have more freedom to interact with neighboring cells, as well as to migrate and interact with the surface receptor or ECM components on the surfaces of the microbeads.

The schematic of microfluidic synthesis of PEG-co-PLL microbeads and 3D cell culture.
A typical microfluidic flow-focusing droplet generator was used to emulsify PEG-co-PLL aqueous pre-gel solution. The droplets were polymerized after flow through the extension channel and exposure to UV irradiation to form soft microbeads. These microbeads (in red color) were modified with decellularized matrix (DCM) components from pancreatic tissue and cultured with β-cells (in green) in direct contact. The inset schematic shows the interface between the microbeads and their neighboring cells. The shell (in dark yellow color) on the microbeads represents the DCM components from pancreatic tissue.
PEG hydrogels are widely used for biomedical applications due to their biocompatibility, high permeability to small molecules, as well as tunable stiffness and chemical compositions. Biofunctional peptides or proteins can be easily introduced to the hydrogel network (i.e. by covalent bonding or copolymerization) while maintaining its general material properties18. We chose PEG-co-PLL polymer hydrogels as our starting material. Cell-adhesive peptides such as RGD and other fibronectin domains and cell surface receptors EphA/EphrinA have both previously been successfully conjugated into plain PEG hydrogels for β-cell encapsulation16. In our approach, the copolymerization of PLL with PEG introduces positive charges on the gel surface, which allows for the absorption of ECM components from pancreatic tissue and provides a handle for receptor conjugation to the gel surface.
Single purified ECM proteins or combinations of various proteins (i.e. collagens, fibronectin, growth factors or laminin, etc.) have been used to modify cell culture substrates to mimic cell-ECM interactions5,7,19,20. Cell-derived matrix coatings from fibroblasts21,22,23,24,25 and Matrigel™ 26 have also been applied to promote cell attachment and improve cell functions. However, these surfaces are not able to recapitulate the complexity of the native microenvironment in terms of the tissue-specific combinations of various ECM components. Recently, decellularization procedures have been optimized to remove cellular components from specific tissues while preserving key molecular components in the ECM27,28,29. Decellularized matrix (DCM) from liver tissue has been developed for 3D culture of hepatocytes and was shown to considerably improve cell viability and proliferation30. Other types of DCM from various tissues and whole organs have also been reported28,31,32. Based on these findings, we hypothesize that absorption of pancreatic DCM components onto the microbeads could similarly emulate the tissue specific native microenvironment for β-cells.
The PEG-co-PLL microbeads were prepared by microfluidic emulsification and UV-polymerization33,34,35, which provides precise control over their size and chemical composition. β-cells are cultured in direct contact with microbeads, which not only mimic the in vivo presentation of cells in the pancreatic islets, but also overcome the diffusion limitations of oxygen and nutrients, as mass transfer can more easily be achieved through the void spaces between the cells and microbeads. Thus, these systems allow the generation of 3D assemblies of functional cells such as β-cells with microgel cell mimics that maintain key cell-cell communications and present components of the native matrix. The result is the sustained maintenance of cell function in vitro and prolonged cell survival. This concept of synthetic “neighbors” is one that we anticipate can be adapted to the maintenance of other functional cell types.
Results
Characterization of decellularization of pancreatic tissue
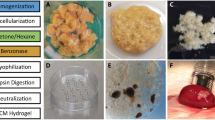
We developed a method to decellularize rat pancreatic tissue by perfusion with Triton X-100, a nonionic detergent that lyses cells and solubilizes cytoplasmic components. After 48 hours perfusion, a translucent acellular tissue was formed (Figure 2a, b). Hematoxylin and eosin (H&E) stain of the resulting tissue revealed no nuclei or cytoplasmic compartments (Figure 2c). The decellularized tissue was lyophilized for three days and milled to powder. The initial weight of the pancreatic tissue from ten adult rats was approximately 65 g, from which was obtained 68.6 mg of lyophilized DCM powder. After 24 hours of pepsin digestion at 37°C, a nearly clear solution of DCM was generated. DCM is a very complex composition of biomolecules, including different types of collagen (type I, IV, etc.), fibronectin, laminin and other proteins and peptides29,36. As shown in the SDS-page image, the solubilized DCM exhibited the presence of lower molecular weight bands similar to those in pancreatic tissue (Figure 2e).

Decellularized matrix (DCM) from pancreatic tissue.
(a) Rat pancreatic tissue was perfused in Triton X-100 aqueous solution. (b) Decellularized pancreatic tissue after 48 hours perfusion. (c) Hematoxylin and eosin (H&E) stained sections of the decellularized pancreatic tissue, as the confirmation of the absence of cells. (d) Powder of DCM after lyophilization. (e) SDS-PAGE of digested pancreatic tissue and solubilized DCM. Note that the pancreatic tissue has a more complex mixture of cellular and extracellular matrix components than DCM, while soluble DCM kept most lower molecular weight bands. The scale bar is 1 cm in a and b, 200 μm in c, respectively.
Microfluidic synthesis of PEG-co-PLL microbeads and 3D culture of β-cells
Free thiol groups were covalently linked onto fusion proteins (EphA5–Fc and EphrinA5–Fc) using 2-iminothiolane-HCl (Traut's reagent)16. Specifically, fusion proteins were dissolved in PBS containing 2 mM EDTA. Traut's reagent (10 μM/mL) was mixed with the fusion protein solution (2 μM/mL) and reacted for 1 hour at room temperature. Excess Traut's reagent was separated from the solution using a desalting column. The pre-gel solution comprises 8 wt% PEGDA (Mw = 1000), 2 mg/mL PLL (Mw = 15,000 ~ 30,000), 1 μM/mL thiolated EphA5-FC and EphrinA5-Fc and 0.5 wt% of photo-initiator HMPP. Thiolated proteins were introduced to the PEG network through thiol-acrylate reactions initiated by UV irradiation37,38. PLL was incorporated into the PEG hydrogel network by physical trapping to yield an interpenetrating network, due to the fact that the molecular dimensions of the PLL are larger than the mesh size of the PEG gel network7,22. The pre-gel solution was injected into a microfluidic flow-focusing droplet generator (Figure 3a)39,40, from which the aqueous stream broke into droplets in mineral oil with uniform sizes. The droplets were partially polymerized for approximately 1 minute in the chip to prevent aggregation and then fully polymerized in the extension tubing for about 4 minutes. The PEG hydrogel networks were formed through two types of reactions: the free radical polymerization of PEG diacrylate and the linking of thiols with acrylate groups38. The size of the microbeads was 21 ± 1 μm with a monodispersity of 2.4% (Figure 3b,c). FITC-labeled PLL was used to confirm a homogeneous distribution of entrapped PLL in the PEG gel network. After the modification of the microbeads with the adsorption of DCM components from aqueous solution, noticeable changes in the size of microbeads were not observed.

Microfluidic synthesis of PEG-co-PLL microbeads and 3D culture of pancreatic β-cells.
(a) Typical optical microscope image of microfluidic generation of PEG-co-PLL droplets. Mineral oil and pre-gel aqueous solution were supplied from side channels and center channel, respectively. (b) Size distribution of the PEG-co-PLL microbeads with mean diameter of 21 ± 1 μm. (c) Polymerized PEG-co-PLL (with or without EphA/EphrinA modification) microbeads were dispersed in solubilized DCM. (d) The size of PEG-co-PLL microbeads in response to the change of flow rates of pre-gel solution. (e) Optical microscope image of pancreatic β-cells cultured with PEG-co-PLL microbeads with a ratio of cells to microbeads of 1:3. The number of cells in each well of a 48-well plate was 1.2 × 105/well. (f) The optical fluorescent image of β-cells with microbeads at day 2, which indicated good contact of cells with microbeads in a 3D manner. The cells were stained with calcein AM (green) and Ethidium homodimer-1 (red) for 30 minutes. The scale bars in a, c, e and f are 100 μm.
Although this work focused on preparation of microbeads with sizes similar to that of β-cells, the microfluidic reactor also can be used to make microbeads with various sizes ranging from 15 to 85 μm. In general, the size of the microbeads is increased by increasing the volumetric flow rate of the pre-gel solution (Figure 3d)34. The number of microbeads in each well was optimized to ensure that the cells and microbeads could form a 3D densely packed structure (Figure 3e). The cells were stained with calcein AM (green color, indication of live cells) and Ethidium homodimer-1 (red color, indication of dead cells) after culturing with microbeads for two days (shown in Figure 3f). We observed single cell, cell-cell cluster and cell-microbead aggregates. Single cells maintained their spherical shape, while cell clusters were observed to spread on the surface or occupy the spaces between several microbeads.
Characterization of PEG-co-PLL microbeads
FT-IR spectrometry was used to qualitatively characterize the chemical composition of microbeads before and after modification of DCM (shown in Figure 4a). PEG-co-PLL samples show combinations of absorption peaks of C-O-C vibration at 1150 cm−1 (from PEG) and N-H stretching at 3400–3250 cm−1 and bending at 1650 cm−1 (from PLL). PEG-co-PLL + DCM show enhanced absorption of the C-H stretching vibration at 2950–2850 cm−1, C = O stretching vibration at 1735 cm−1, N-H bending vibration at 1640 cm−1 and C-H bending vibration at 1470 and 1360 cm−1, mostly from different types of collagen41, which were found in the DCM spectra.

Characterization of PEG-co-PLL + DCM microbeads.
(a) FT-IR spectra of PEG-co-PLL + DCM microbeads, PEG-co-PLL microbeads and DCM. (b), (c) SEM images of lyophilized PEG-co-PLL and PEG-co-PLL + DCM microbeads, respectively. The insets high magnification images show the surface morphologies of the microbeads. The scale bar of the inset images is 2 μm.
Figures 4b and c show the SEM images of lyophilized PEG-co-PLL and PEG-co-PLL + DCM microbeads, respectively. Compared to the size of the wet microbeads, the sizes of the lyophilized microbeads decreased from 21 μm to 15 μm. No significant size difference was observed for these two types of microbeads. Clumps/clusters of PEG-co-PLL microbeads were formed during the freeze-drying process, probably due to the crystallization of water in the polymer network. In comparison, PEG-co-PLL + DCM microbeads exhibited much less aggregation. Furthermore, the surface morphologies of PEG-co-PLL and PEG-co-PLL + DCM microbeads are quite different after lyophilization: PEG-co-PLL microbeads have a very smooth surface, indicating the homogeneous structure of the hydrogel network; PEG- co-PLL + DCM microbeads have a bumpy and heterogeneous surface. This observation suggests that a thin shell of polymer (mostly a mixture of collagen and other negatively charged proteins) remains adsorbed to the surface of the PEG- co-PLL microbeads after the modification with DCM.
Live/dead cell viability test on β-cells cultured with various types of microbeads
β-cells were cultured with four types of PEG-co-PLL microbeads, specifically, PEG-co-PLL microbeads (Beads only), EphA/EphrinA fusion protein modified PEG-co-PLL Beads (Beads + EphA/EphrinA), PEG-co-PLL microbeads absorbed with DCM (Beads + DCM) and EphA/EphrinA fusion protein modified PEG-co-PLL Beads absorbed with DCM (Beads + EphA/EphrinA + DCM). Control experiments were carried out on cells plated in regular polystyrene culture plates (non-coating). Cell viability at days 2, 7 and 21 was tested using a Live/Dead viability/cytotoxicity kit for from Invitrogen.
At day 2, we did not observe significant differences in cell viability among all five conditions. Cells were evenly distributed on a polystyrene substrate (as a control) or in the microbeads in the cell media (as shown in Figure 3f). Fluorescent images were taken and used as references for quantitative analysis. At day 7, we observed that approximately 33% of cells cultured in the non-coated 2D microwell control were not viable, while cells cultured with microbeads maintained over 90% survival for all four types of microbeads. Among these types of microbeads, cells cultured in Beads + EphA/EphrinA + DCM showed the highest survival at 95%, which is slightly higher than those in other three types of microbeads (not statistically significant). Cells cultured on 2D substrates spread out on the surface, while cells cultured in 3D microbeads maintain their spherical shape. We also observed that cell clusters formed in in the 3D culture. At day 21, only 14% of cells were viable in the uncoated 2D culture. For the cells cultured in 3D with microbeads, we observed enhanced cell viability of 28%, 17%, 26% and 69% for the conditions of Beads only, Beads + EphA/EphrinA, Beads + DCM and Beads + EphA/EphrinA + DCM, respectively. Most surviving cells were spherical in shape and some formed aggregations with cell-cell or cell-microbead contacts. In the cells cultured with Beads + EphA/EphrinA + DCM, we observed large populations of 3D cluster aggregates of β-cells and microbeads (shown in Figure 5c), comprised of multiple live cells and a range of 5 to 40 microbeads.

Live/dead cell viability assay of pancreatic β-cells cultured with various PEG-co-PLL microbeads at day 7 and day 21.
(a) Optical fluorescent images of β cell cultured with various types of PEG-co-PLL microbeads. Cells cultured conventional polystyrene substrate was used as control (non-coating). The cells were stained with calcein AM (green) and Ethidium homodimer-1 (red). (b) Quantitative analysis of live/dead cell viability of cells cultured in the various types of microbeads. n = 4. The error bars represent the s.d. of three measurements. (c) Fluorescent image of aggregation of β-cells (green color) and the microbeads (dark color, indicated by the arrow) underneath the cells. The scale bar is 200 μm in a and 100 μm in c, respectively.
Size distribution of the aggregates formed by β-cells with various types of microbeads
Aggregates of cell/cell and cell/microbead were observed in the first two to four days. Cells cultured on the 2D polystyrene substrate (2D) spread out on the surface, while cells cultured with microbeads primarily maintain spherical morphology. We analyzed the size distribution of cells and aggregates for day 7 and day 21 (shown in Figure 6). Three size ranges used here were 1–100, 100–400 and > 400 μm2, representing single cell, small aggregates and large aggregates, respectively. At day 7, the average cross-sectional areas of the aggregates under 3D conditions are approximately 210 ± 20 μm2, which is slightly larger than that on 2D surface (170 ± 20 μm2). However, we did not observe significant differences among the cells cultured with four different microbead conditions (3D). The size distributions of those aggregates are also similar for three ranges with 45 ± 2% in 1–100 μm2, 39 ± 2% in 100–400 μm2 and 16 ± 2% in > 400 μm2, respectively. At day 21, compared to the cells cultured in 2D conditions, the cell populations in the condition of Bead + EphA/EphrinA + DCM have significantly fewer single cells and more aggregates in the size range of > 400 μm2. For the other three 3D conditions (Beads only, Beads + EphA/EphrinA and Beads + DCM), we also observed this trend, but it was not substantial.

Size distribution of the aggregates formed by β-cells and with various types of PEG-co-PLL microbeads at day 21.
The red lines indicate the typical size distribution of those aggregates at day 7.
β-cell specific genes expression and glucose stimulated insulin secretion (GSIS)
Quantitative PCR (qPCR) was used to monitor the expression of several important β-cell specific genes, specifically, v-Maf musculoaponeurotic fibrosarcoma oncogene homologue A (MafA), insulin 2 (Ins2) and pancreatic duodenal homeobox factor (Pdx1). After 21 days β-cells cultured with Beads + EphA/EphrinA + DCM have 19%, 68% and 45% increased expression of MafA, Ins2 and Pdx1 genes, respectively, over cells cultured on tissue culture plastic alone. Conversely, cells cultured with only Beads, Beads + EphA/EphrinA, or Beads + DCM exhibited down regulation for MafA, Ins2 and Pdx1 genes relative to TCPS cultured cells, −55–−42%, −37–−23% and −59–−51% respectively (Figure 7a). The glucose stimulated insulin response (GSIS) for β-cells cultured with Beads + EphA/EphrinA + DCM was significantly improved, over 55% closer to baseline levels as compared to cells cultured on TCPS only after one week. Improved GSIS for cells cultured with Beads + EphA/EphrinA + DCM relative to TCPS was observed out to 21 days. At 21 days β-cells grown on TCPS demonstrated a negative response to high-glucose treatment, while those cultured in contact with Beads + EphA/EphrinA + DCM retained a positive response, secreting 40% more insulin when exposed to high-glucose conditions. These results demonstrate the importance of cell–cell contact and cell-matrix interactions and how these are critical in maintaining normal glucose responsive insulin secretion.

Function of β-cells cultured in various conditions.
(a) qPCR analysis of β-cell specific gene (MafA, Ins2 and Pdx1) expression in cells cultured with various types of PEG-co-PLL microbeads at 21 days. (b) Glucose stimulated insulin secretion (GSIS) of β-cells cultured with Beads + EphA/EphrinA + DCM or in 2D Non-coating (TCPS) surface. Cells were cultured in a 48-well microplate, with approximately 1.2 × 105 cells/well. In the microbead containing conditions, 3.6 × 105 microbeads/well were used. n = 4. The error bars represent the s.d. of three measurements.
Population change of β-cells in 2D and 3D conditions
To follow the dynamics of the decline in β-cell population, the dissociated cells were infected with two lentiviruses that express fluorescent proteins helpful for visualization of β-cells (Figure 8, single fluorescent channel images and bright-field images are included in Supplementary Information). Specifically, cells were engineered to express a blue fluorescent protein constitutively and a red fluorescent protein as a function of insulin promoter activity. As shown in Figure 8a, approximately 50% of the cells were infected by the lentivirus; insulin-producing β-cells express both red and blue fluorescent proteins, while non-insulin-producing β-cells and other types of cells express only blue fluorescent proteins. For all cases, blue fluorescence did not decrease significantly during the course of the experiments, showing that our heterologous gene constructs were not silenced epigenetically. However, when β-cells were cultured on a standard 2D uncoated culture plate surface, red fluorescence decreased after 7 days (Figure 8b) and almost completely disappeared after 21 days (Figure 8c), suggesting that cultured β-cells lost their function. In comparison, when cells were cultured with Beads + EphA/EphrinA + DCM, red fluorescence was still strongly visible after 7 and 21 days (Figure 8d and 8e).

Population change of β-cells in 2D and 3D conditions.
Fluorescent microscopy images of lentivirally infected β-cell and cell aggregates cultured at non-coating 2D surface (a for after infection, b for day 7 and c for day 21, respectively) and cultured with Beads + EphA/EphrinA + DCM (d for day 7 and e for day 21, respectively). Cells were double infected with a lentivirus encoding hIns(3×)_mKate-2A-Puro that expresses a red fluorescent protein mKate (along with translationally fused Puromycin resistance protein) depending on insulin promoter hIns(3×) activity and a lentivirus encoding hEF1a_EBFP2 that constitutively expresses blue fluorescent protein EBFP2 from hEF1a promoter. Puromycin selection was not used in this experiment. Cells were cultured in a 48-well microplate, with approximately 6 × 104 cells/well initially. In the microbeads conditions, 3.6 × 105 microbeads/well were used. Insets show a zoom-in of representative cells, with a scale bar of 20 μm.
Discussion
Both type 1 and type 2 diabetes are characterized by the loss or dysfunction of pancreatic β-cells42. Therefore, the formation of functional new β-cells is considered to be an ultimate cue for diabetes therapy. Strategies for increasing the β-cell mass include inhibiting β-cell apoptosis and stimulating β-cell replication of original cells, as well as regeneration from embryonic stem/progenitor cells42,43. In both cases, creating an ex vivo microenvironment for β-cells is critical to enhance their survival and maintain their phonotype.
Previous studies have demonstrated that cell–cell and cell–ECM interactions are the important for the survival and function of pancreatic β-cells7,12,16. The understanding of critical cell–cell and cell–ECM interactions is the key to engineering biomimetic microenvironments that can recapitulate the 3D organization of β-cells in the pancreatic tissue. For in vitro cell culture, increasing cell-seeding density is a common method to increase the possibility of cell-cell contact and decreases the distance between cells to enhance paracrine signaling18,44. However, it is not practical due to the high cost for the isolation of primary β-cells. As an alterative approach, cell-cell interactions were stimulated by the encapsulation of cells into polymer hydrogels that conjugated with cell surface receptors EphA and EphrinA16. For cell-ECM interactions, specific or combination of ECM proteins can be selectively conjugated onto synthetic PEG hydrogels; as a result, they have been widely used for the study of encapsulation of cells for mimicking the cell-ECM interactions on promoting the survival of pancreatic β-cells. Efforts include immobilization of glucagon- like peptide 1 (GLP-1) to enhance β-cell encapsulation20, modification of RGDs to improve cell attachment6, conjugation of affinity peptides to sequester cytokine45 to improve immunoisolation of hydrogels. However, mostly of the findings from current work were carried out from MIN6 insulinoma cell line, not from primary β-cells. Furthermore, these hydrogel systems were not able to reconstruct the native microenvironment of the pancreatic tissue-specific ECM. How to enhance the survival and stability of primary β-cells remains a critical challenge.
Recently, extensive efforts have contributed to the development of decellularized tissue or organs, which have shown success in removing cellular components and preserving important components in the ECM from heart, liver, muscle and other tissue28,30,31,32. Similar to those approaches, decellularized pancreatic tissue contains key ECM components, which provides “pseudo-native” environment for in vitro culture of β-cells. As the first step of this work, we optimized a method to prepare DCM from pancreatic tissue. Specifically, The alkaline solution of NH4OH was used to solubilize cytoplasmic components and disrupt nucleic acids and the non-ionic detergent Triton X-100 was used to disrupt lipid-lipid and lipid-protein interactions, while leaving protein-protein interaction intact27. After decellularization, all residual chemicals are carefully removed to avoid any adverse host tissue response to the chemicals. The solution of the digested pancreatic DCM is first tested on polystyrene substrates. Rat primary β-cells cultured on a DCM modified substrate showed improvements on cell viability and stability over two weeks (unpublished results).
Then, EphA5–Fc and EphrinA5-FC modified PEG-co-PLL microbeads were prepared from droplets generated by a microfluidic reactor. Positively charged PLL in the polymer network ensured weak positive charges on the surfaces of the microbeads. Then a solution of DCM with negatively charged ECM components was absorbed onto the microbead surface and created a thin shell of extracellular matrix, as confirmed by FT-IR results and SEM images shown in Figure 4. When dispersed β-cells were cultured among these microbeads in cell media, cells and beads created close-packing 3D structures that include direct contact of cell-to-cell and cell-to-ECM.
Several previous studies have shown that without cell–ECM interactions, dissociated β-cells cannot survive when they are encapsulated in synthetic hydrogels20,46. G. C. Weir and colleagues have demonstrated that small β-cell re-aggregates were able to survive and function better than intact islets within alginate microcapsules, mostly due to the fact of sufficient oxygen transport and enhanced cell-cell contact47. Recently, the Anseth group revealed a type of cell-laden hydrogel that mimics the synergistic effect of cell-ECM and cell-cell interactions and the dissociated β-cells in the hydrogel can survive at densities over 1 × 106 cells/mL16. However, the majority of encapsulated β-cells underwent apoptosis in ten days, mostly due to nonsufficient cell-ECM and cell-cell interactions. In our approach, the presentation of β-cells in the microbeads mimicked the 3D organization and aggregation of cells in the pancreatic islets. Cells and microbeads were in close contact, which initiated the aggregation of cells with other beta cells and with microbeads. As shown in Figure 5, cells were present in all four 3D conditions; and no significant difference among them at day 7, suggested that aggregation (as increasing the local cell-seeding density) of cells enhanced β-cell survival by providing sufficient cell–cell interactions. This is in good agreement with the work reported by previous researchers18,47. We also found that the size and size distributions of the aggregates are very similar in all four 3D conditions.
At day 21, β-cells cultured in the 3D synthetic “neighbor” microbeads structure had better viability as well. More importantly, cells in the Beads + EphA/EphrinA + DCM condition had over five fold higher viable cells comparing to cells on 2D surface, while other 3D conditions (Beads only, Beads + EphA/EphrinA and Beads + DCM) also increase the cell viability. We further checked the size and size distributions of the cell aggregates at day 21 (shown in Figure 6). The better survival rate for the Beads + EphA/EphrinA + DCM cells was most likely due to the larger sizes of the aggregates (fewer single cells) and synergistic effects of both cell-to-cell and cell-to-ECM interactions. Furthermore, we checked the expression of β-cell specific genes for the survivor cells at day 21, as shown in Figure 7a. MafA, Ins2 and Pdx1 are typical genes that undergo downregulation when β-cells lose their identity and insulin producing ability48,49. Compared to the cells on the 2D control surface, we observed 20–70% upregulation of those genes for the cells cultured in the Beads + EphA/EphrinA + DCM condition, suggesting better β-cell function for the cells in this 3D microenvironment. Downregulation of these three genes for the other three 3D conditions (Beads only, Beads + EphA/EphrinA and Beads + DCM) also indicated that some populations of survived cells lose the β-cell identity due to the lack of above synergistic effects. Additionally, as shown in Figure 7(b), cells cultured with Beads + EphA/EphrinA + DCM retained a positive GSIS response, an indicator of proper β-cell function out to 21 days in culture. We also demonstrated sustained insulin promoter activity in cells cultured with Beads + EphA/EphrinA + DCM over 21 days which was lost in cells cultured in 2D Non-coating surface (Figure 8). Our studies suggest that increased cell survival and function with microbead-based microenvironment may be attributed to: (i) increased local cell density for actual cell–cell contact, (ii) efficient cell-cell and cell-ECM interactions between the beta cells and microbead synthetic neigbors, (iii) ECM from native pancreatic tissue, (iv) synergistic effects of both cell-to-cell and cell-to-ECM interactions and (v) elimination of mass transport limitations of oxygen and nutrients in the 3D microenvironment.
In summary, we presented the approach of using close-packed PEG-co-PLL microbeads to create a biomimetic 3D microenvironment to improve the survival and function of primary pancreatic β-cells. We demonstrated that selective modification of these microbeads with cell surface cadherin receptors and pancreatic tissue specific extracellular matrix can provide sufficient native-like cell-cell and cell-ECM interactions, which initiate the formation of cell-cell-bead aggregates and create synergistic effects for comprehensive interactions between real β-cells and the microbeads (“pseudo β-cells”). Primary pancreatic β-cells cultured in this platform had a five-fold enhanced rate of survival and had significantly improved expression of β-cell specific genes and glucose stimulated insulin secretion for up to 21 days in culture.
Our microbead-based microenvironment provides a new method to maintain primary β-cell phenotype and function in vitro for clinical applications, which has been a long-standing obstacle for diabetes therapy. Following the dynamics of the decline in β-cell mass might provide insight to the pathologic mechanisms of both Type 1 and Type 2 diabetes50. This technique could be applied to evaluate interventional efficacies as well as improving our understanding of the effects of medication and glycemic control on β-cell mass. Our approach also provides a possible strategy for directing in vitro β-cell expansion by incorporating proper growth factors into the microbead-based 3D culture system. Furthermore, taking advantage of microfluidic synthetic techniques, polymer microbeads with controlled physical, chemical and biological properties (i.e. size, shape, stiffness, chemical composition, conjugation of different types of bioactive ligands, etc.) can be easily achieved. Together with a tissue-specific extracellular matrix, microbead-based cell culture platforms can open a variety of opportunities for the study of complex biological problems for other types of cells.
Methods
The preparation of decellularized matrix (DCM) and DCM coating solution
All procedures were in compliance with the Federal Regulation on Animal Care and Harvard Medical School animal use protocol, following all the ethical guidelines for experimental animals. Maintenance of mouse colonies and all experiments were conducted in accordance with the Guide for the Use and Care of Laboratory Animals and approved by the Harvard Medical Area Standing Committee on Animals. Pancreatic tissue was harvested from adult rats and perfused with D-PBS. Connective tissue and fat was removed. The tissue was then cut into ~ 1 cm3 pieces and decellularized. Specifically, the tissue was rinsed with deionized water and then stirred in 1 wt% aqueous solution of Triton X-100 with 0.01 M ammonium hydroxide (NH4+OH−) for 48 hours. The decellularized pancreatic tissue was rinsed with deionized water to completely remove the detergent. A sample of decellularized matrix was frozen and sectioned into 10 mm slices and stained with hematoxylin and eosin (H&E) to confirm the absence of cells. The decellularization tissue was lyophilized for 48 hours and milled into fine-grained powder and stored at −20°C. The milled powder of decellularized tissue was solubilized through enzymatic digestion29. Pepsin (SIGMA, St. Louis, MO) was dissolved in 0.01 M hydrochloric acid (HCl) to make 1 mg/mL solution. Approximately 10 mg of the DCM was digested in 1 mL of pepsin solution at 37°C under constant shaking. After 24 hours, the pH of the digestion solution was adjusted to 7.4 with 0.1 M sodium hydroxide (NaOH) solution and diluted with 10 × D-PBS. The final concentration of DCM in the PBS solution is 4 mg/mL.
Thiolation of receptors and preparation of PEG-co-PLL solutions
Free thiol groups were introduced onto fusion proteins (EphA5–Fc and EphrinA5–Fc) using 2-iminothiolane-HCl (Traut's reagent) according to the method reported by the Anseth group16. Specifically, fusion proteins were dissolved in PBS containing 2 mM EDTA. Traut's reagent (10 μM) was mixed with the fusion protein solution (2 μM and reacted for 1 hour at room temperature. Excess Traut's reagent was separated from the thiolated proteins solution using a desalting column (Zeba™ Spin, 40 K, ThermoFisher Scientific). The pre-gel aqueous solution comprises the following ingredients: 8 wt% poly(ethylene glycol) dimethylacrylate (PEGDMA: Mw = 1000, Polyscience), 2 mg/mL poly-L-lysine hydrobromide (PLL, Mw = 15,000 ~ 30,000), 0.2 μM thiolated EphA5-FC and EphrinA5-Fc and 0.5 wt% of photo-initiator 2-hydroxy-2-methylpropiophenone (HMPP).
Microfluidic synthesis of PEG-co-PLL microbeads and surface modification
Masters for preparing microfluidic reactor were prepared with an SU-8 photoresist in bas-relief on silicon wafers. The microfluidic device was fabricated in poly(dimethylsiloxane) (PDMS) by using a standard soft-lithography method51. The thickness of the microochanel is approximately 80 μm. PEG-co-PLL microbeads with mono-dispersed size distribution were carried out in the microfluidic reactors35,52. Briefly, two immiscible liquids, an aqueous PEG-co-PLL pre-gel solution and a mineral oil solution with 2 wt% surfactant Span80 and 0.5 wt% photo-initiator HMPP were supplied to the microchannels using two digitally controlled syringe pumps (Harvard Apparatus PHD 2000, U.S.A.). After emulsification, the PEG-co-PLL droplets were flowed in extension tubing and exposed to UV irradiation for 5 minutes. A Zeiss Axioskop 2 optical and fluorescent microscope and a high-speed digital camera were used to acquire images. Various flow rates of oil phase and pre-gel phase were used to prepare microbeads with designed sizes. After polymerization, the microbeads were separated from oil phase by centrifugation (1500 rpm, 5 minutes), washed with 0.2 wt% of Triton X-100 aqueous solution, then washed with DI water to remove oil, surfactant and other residues. To modify the microbeads with DCM, the beads were added into 4 wt% DCM solution and incubated at 37°C for four hours. Then the microbeads were washed with cell media for several times and stored in cell media for future use. The monodispersity of the microbeads was defined as standard deviation divided by the mean size.
Characterization of PEG-co-PLL microbeads
PEG-co-PLL and PEG-co-PLL + DCM microbeads were lyophilized for 72 hours. The chemical compositions of these microbeads were analyzed by Fourier-transform infrared (FT-IR) (Nexus 870, Thermo Scientific) spectroscopy for the dry beads. Scanning electron microscopy (SEM) (JEOL JSM-6060) was also used to visualize the surface morphology of the dry microbeads.
Cell culture and characterization of DCM
Rat pancreatic islets were isolated as previous described53, then the islets were dissociated into single cell mixture (mostly β-cells) using 0.05% trypsin EDTA solution16 and no further purification was performed. The cell media is low glucose DMEM medium containing 2% fetal bovine serum (FBS), 1% penicillin-streptomycin. Medium was changed every 2–3 days. Typically, cells with microbeads were cultured in a 48-well microplate (BD Biosciences, San Jose, CA), with approximately 1.2 × 105 cells/well and 3.6 × 105 microbeads/well. We generated four types of microbeads, specifically, PEG-co-PLL microbeads (Beads only), EphA/EphrinA fusion protein modified PEG-co-PLL microbeads (Beads + EphA/EphrinA), PEG-co-PLL microbeads absorbed with DCM (Beads + DCM) and EphA/EphrinA fusion protein modified PEG-co-PLL microbeads absorbed with DCM (Beads + EphA/EphrinA + DCM). Cells cultured without microbeads in the microwells were used as a control (non-coating). Solubilized DCM was analyzed by SDS-PAGE and compared to lysed pancreatic tissue. Solutions were run on a Tris–HCl, 12% polyacrylamide gel (Bio-Rad Laboratories, Inc, Hercules, CA) in Tris/Glycine/SDS buffer (Fisher Scientific Inc, Hanover Park, IL), with 80 mM reducing agent Dithiothreitol (DTT) (Invitrogen, Carlsbad, CA). Gel electrophoresis was performed in an XCell Surelock MiniCell (Invitrogen), compared to a broad range standard and stained with Imperial Protein Stain (Pierce, Rockford, IL).
Cell viability in various types of microbeads
Cell viability was determined using a LIVE/DEAD® viability kit. Cells cultured with various types of mirobeads were placed in D-PBS containing calcium AM and ethidium homodimer according to manufacturer's staining protocol for at least 45 minutes. Briefly, live/dead cells are distinguished by intracellular esterase activity, which was determined by the enzymatic conversion of the virtually nonfluorescent dye calcein AM to the fluorescent calcein. The polyanionic dye calcein was retained within live cells, producing an intense uniform green fluorescence in live cells (ex/em ~ 495 nm/~515 nm). EthD-1 enters cells with damaged membranes and undergoes a 40-fold enhancement of fluorescence upon binding to nucleic acids, thereby producing a bright red fluorescence in dead cells (ex/em ~ 495 nm/~635 nm). EthD-1 is excluded by the intact plasma membrane of live cells.
Analysis of the size of cell-cell-beads aggregates
The size and size distributions of the aggregates formed by β-cells and various types of microbeads were quantified by using ImageJ. Briefly, the florescent images taken from the Live/Dead® viability assay were split into green and red channels and only the live cells and aggregates in the green channel were analyzed. The images were converted to threshold format to count the numbers and determine the area of the cells and aggregates by the software ImageJ. We defined three groups of size ranges as 1–100, 100–400 and > 400 μm2, representing single cell, small aggregates and large aggregates, respectively.
References
Walker, G. M., Zeringue, H. C. & Beebe, D. J. Microenvironment design considerations for cellular scale studies. Lab Chip 4, 91–97 (2004).
Choi, C. K., Breckenridge, M. T. & Chen, C. S. Engineered materials and the cellular microenvironment: a strengthening interface between cell biology and bioengineering. Trends Cell Biol. 20, 705–714 (2010).
Huh, D., Hamilton, G. A. & Ingber, D. E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 21, 745–754 (2011).
Bhatia, S. N., Balis, U. J., Yarmush, M. L. & Toner, M. Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 13, 1883–1900 (1999).
Bryant, S. J. & Anseth, K. S. Controlling the spatial distribution of ECM components in degradable PEG hydrogels for tissue engineering cartilage. J Biomed Mater Res Pt A 64A, 70–79 (2003).
Kinney, M. A., Saeed, R. & McDevitt, T. C. Systematic analysis of embryonic stem cell differentiation in hydrodynamic environments with controlled embryoid body size. Integr. Biol. 4, 641–650 (2012).
Weber, L. M., Hayda, K. N. & Anseth, K. S. Cell-matrix interactions improve beta-cell survival and insulin secretion in three-dimensional culture. Tissue Eng Pt A 14, 1959–1968 (2008).
Flaim, C. J., Teng, D., Chien, S. & Bhatia, S. N. Combinatorial signaling microenvironments for studying stem cell fate. Stem Cells Dev. 17, 29–39 (2008).
Lutolf, M. P. & Hubbell, J. A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 23, 47–55 (2005).
Zhang, C. et al. The controlled presentation of TGF-beta 1 to hepatocytes in a 3D-microfluidic cell culture system. Biomaterials 30, 3847–3853 (2009).
Pisania, A. et al. Quantitative analysis of cell composition and purity of human pancreatic islet preparations. Lab. Invest. 90, 1661–1675 (2010).
Jain, R. & Lammert, E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes Metab. 11, 159–167 (2009).
Kohen, E., Kohen, C. & Rabinovitch, A. cell-to cell communication in rat pancreatic-islet monolayer-cultures is modulated by agents affecting islet-cell secretory activity. Diabetes 32, 95–98 (1983).
Luther, M. J. et al. Cell-to-cell contact influences proliferative marker expression and apoptosis in MIN6 cells grown in islet-like structures. Am J Physiol-Endo and M. 288, E502–E509 (2005).
Konstantinova, I. et al. EphA-ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 129, 359–370 (2007).
Lin, C. C. & Anseth, K. S. Cell-cell communication mimicry with poly(ethylene glycol) hydrogels for enhancing beta-cell function. Proc. Natl. Acad. Sci. U. S. A. 108, 6380–6385 (2011).
Johnson, A. S., Fisher, R. J., Weir, G. C. & Colton, C. K. Oxygen consumption and diffusion in assemblages of respiring spheres: Performance enhancement of a bioartificial pancreas. Chem. Eng. Sci. 64, 4470–4487 (2009).
Lin, C. C. & Anseth, K. S. PEG Hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm. Res. 26, 631–643 (2009).
Benoit, D. S. W., Durney, A. R. & Anseth, K. S. The effect of heparin-functionalized PEG hydrogels on three-dimensional human mesenchymal stem cell osteogenic differentiation. Biomaterials 28, 66–77 (2007).
Lin, C. C. & Anseth, K. S. Glucagon-like peptide-1 functionalized PEG hydrogels promote survival and function of encapsulated pancreatic beta-cells. Biomacromolecules 10, 2460–2467 (2009).
Cooper, S. T. et al. C2C12 co-culture on a fibroblast substratum enables sustained survival of contractile, highly differentiated myotubes with peripheral nuclei and adult fast myosin expression. Cell Motil. Cytoskeleton 58, 200–211 (2004).
Luo, Y., Kobler, J. B., Zeitels, S. M. & Langer, R. Effects of growth factors on extracellular matrix production by vocal fold fibroblasts in 3-dimensional culture. Tissue Eng. 12, 3365–3374 (2006).
Radisic, M. et al. Pre-treatment of synthetic elastomeric scaffolds by cardiac fibroblasts improves engineered heart tissue. J Biomed Mater Res Pt A 86A, 713–724 (2008).
Ueno, H. et al. Evaluation effects of chitosan for the extracellular matrix production by fibroblasts and the growth factors production by macrophages. Biomaterials 22, 2125–2130 (2001).
VanWinkle, W. B., Snuggs, M. B. & Buja, L. M. Cardiogel: A biosynthetic extracellular matrix for cardiomyocyte culture. In Vitro Cell Dev-An 32, 478–485 (1996).
Kleinman, H. K. & Martin, G. R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 15, 378–386 (2005).
Gilbert, T. W., Sellaro, T. L. & Badylak, S. F. Decellularization of tissues and organs. Biomaterials 27, 3675–3683 (2006).
Song, J. J. & Ott, H. C. Organ engineering based on decellularized matrix scaffolds. Trends Mol. Med. 17, 424–432 (2011).
DeQuach, J. A. et al. Simple and high yielding method for preparing tissue specific extracellular matrix coatings for cell culture. Plos One 5, e13039 (2010).
Lang, R. et al. Three-dimensional culture of hepatocytes on porcine liver tissue-derived extracellular matrix. Biomaterials 32, 7042–7052 (2011).
Crapo, P. M. et al. Biologic scaffolds composed of central nervous system extracellular matrix. Biomaterials 33, 3539–3547 (2012).
Wolf, M. T., Daly, K. A., Reing, J. E. & Badylak, S. F. Biologic scaffold composed of skeletal muscle extracellular matrix. Biomaterials 33, 2916–2925 (2012).
Li, W., Greener, J., Voicu, D. & Kumacheva, E. Multiple modular microfluidic (M3) reactors for the synthesis of polymer particles. Lab Chip 9, 2715–2721 (2009).
Li, W. et al. Multi-step microfluidic polymerization reactions conducted in droplets: The internal trigger approach. J. Am. Chem. Soc. 130, 9935–9941 (2008).
Nie, Z. H., Xu, S. Q., Seo, M., Lewis, P. C. & Kumacheva, E. Polymer particles with various shapes and morphologies produced in continuous microfluidic reactors. J. Am. Chem. Soc. 127, 8058–8063 (2005).
Stendahl, J. C., Kaufman, D. B. & Stupp, S. I. Extracellular matrix in pancreatic islets: relevance to scaffold design and transplantation. Cell Transplant. 18, 1–12 (2009).
Rydholm, A. E., Bowman, C. N. & Anseth, K. S. Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials 26, 4495–4506 (2005).
Salinas, C. N. & Anseth, K. S. Mixed mode thiol-acrylate photopolymerizations for the synthesis of PEG-peptide hydrogels. Macromolecules 41, 6019–6026 (2008).
Anna, S. L., Bontoux, N. & Stone, H. A. Formation of dispersions using "flow focusing" in microchannels. Appl. Phys. Lett. 82, 364–366 (2003).
Ward, T., Faivre, M. & Stone, H. A. Drop production and tip-streaming phenomenon in a microfluidic flow-focusing device via an interfacial chemical reaction. Langmuir 26, 9233–9239 (2010).
Belbachir, K., Noreen, R., Gouspillou, G. & Petibois, C. Collagen types analysis and differentiation by FTIR spectroscopy. Anal. Bioanal. Chem. 395, 829–837 (2009).
Bonner-Weir, S. & Weir, G. C. New sources of pancreatic beta-cells. Nat. Biotechnol. 23, 857–861 (2005).
Efrat, S. In vitro expansion of human beta cells. Diabetologia 51, 1338–1339 (2008).
La Flamme, K. E., LaTempa, T. J., Grimes, C. A. & Desai, T. A. The effects of cell density and device arrangement on the behavior of macroencapsulated beta-cells. Cell Transplant. 16, 765–774 (2007).
Lin, C. C., Metters, A. T. & Anseth, K. S. Functional PEG-peptide hydrogels to modulate local inflammation induced by the pro-inflammatory cytokine TNF alpha. Biomaterials 30, 4907–4914 (2009).
Su, J., Hu, B. H., Lowe, W. L., Kaufman, D. B. & Messersmith, P. B. Anti-inflammatory peptide-functionalized hydrogels for insulin-secreting cell encapsulation. Biomaterials 31, 308–314 (2010).
O'Sullivan, E. S. et al. Rat islet cell aggregates are superior to islets for transplantation in microcapsules. Diabetologia 53, 937–945 (2010).
Pechhold, S. et al. Transcriptional analysis of intracytoplasmically stained, FACS-purified cells by high-throughput, quantitative nuclease protection. Nat. Biotechnol. 27, 1038–U1106 (2009).
Murtaugh, L. C. Pancreas and beta-cell development: from the actual to the possible. Development 134, 427–438 (2007).
Weir, G. C. & Bonner-Weir, S. Islet β-cell mass in diabetes and how it relates to function, birth and death. Ann. N.Y. Acad. Sci. 1281, 92–105 (2013).
Xia, Y. N. & Whitesides, G. M. Soft lithography. Annu. Rev. Mater. Sci. 28, 153–184 (1998).
Garstecki, P., Stone, H. A. & Whitesides, G. M. Mechanism for flow-rate controlled breakup in confined geometries: A route to monodisperse emulsions. Phys. Rev. Lett. 94, 164501–164504 (2005).
Ma, M. et al. Core–shell hydrogel microcapsules for improved islets encapsulation. Adv. Healthcare Mater. 2, 667–672 (2013).
Acknowledgements
We acknowledge financial support by the NSF Emergent Behavior of Integrated Cellular Systems (EBICS) Center. The authors also would like to thank the Koch Institute for Integrative Cancer Research and Institute for Soldier Nanotechnologies at MIT for providing resources (facilities, funding) central to the completion of this work. WL would like to acknowledge National Sciences and Engineering Research Council (N.S.E.R.C.) Canada for a postdoctoral fellowship. The authors acknowledge the service to the MIT community of the late Sean Collier.
Author information
Authors and Affiliations
Contributions
W.L., S.L. and P.T.H. conceived and designed the experiments. W.L., S.L., M.M. and S.M.K. carried out the experiments and analyzed the data. J.P. performed the experiments on decellularized matrix. P.G. performed the experiments on cell infection. W.L. and P.T.H. wrote the manuscript. D.G.A., R.W. and R.T.L. contributed in the interpretation and discussion of the experimental results. All of the co-authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Beta-cells
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, W., Lee, S., Ma, M. et al. Microbead-based biomimetic synthetic neighbors enhance survival and function of rat pancreatic β-cells. Sci Rep 3, 2863 (2013). https://doi.org/10.1038/srep02863
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02863
This article is cited by
-
Engineering and Characterization of a Biomimetic Microchip for Differentiating Mouse Adipocytes in a 3D Microenvironment
Pharmaceutical Research (2022)
-
Tissue repair and regeneration with endogenous stem cells
Nature Reviews Materials (2018)
-
3D-Models of Insulin-Producing β-Cells: from Primary Islet Cells to Stem Cell-Derived Islets
Stem Cell Reviews and Reports (2018)
-
Carboxybetaine-modified succinylated chitosan-based beads encourage pancreatic β-cells (Min-6) to form islet-like spheroids under in vitro conditions
Journal of Materials Science: Materials in Medicine (2018)
-
Physics Models of Plasmonics: Single Nanoparticle, Complex Single Nanoparticle, Nanodimer, and Single Nanoparticle over Metallic Thin Film
Plasmonics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
