
Abstract
We report a facile and green method to synthesize a new type of catalyst by coating Pd nanoparticles (NPs) on reduced graphene oxide (rGO)-carbon nanotube (CNT) nanocomposite. An rGO–CNT nanocomposite with three-dimensional microstructures was obtained by hydrothermal treatment of an aqueous dispersion of graphene oxide (GO) and CNTs. After the rGO–CNT composites have been dipped in K2PdCl4 solution, the spontaneous redox reaction between the GO–CNT and PdCl42− led to the formation of nanohybrid materials consisting rGO–CNT decorated with 4 nm Pd NPs, which exhibited excellent and stable catalytic activity: the reduction of 4-nitrophenol to 4-aminophenol using NaBH4 as a catalyst was completed in only 20 s at room temperature, even when the Pd content of the catalyst was 1.12 wt%. This method does not require rigorous conditions or toxic agents and thus is a rapid, efficient and green approach to the fabrication of highly active catalysts.
Similar content being viewed by others


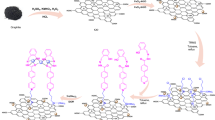
Introduction
Noble metal nanoparticles (NPs) have attracted considerable attention because of their excellent catalytic properties and their potential applications, which include organic synthesis, fuel cells and environmental protection1,2,3. Because the high surface energy of NPs usually causes extensive aggregation4,5,6,7, stabilizers and supporting materials have typically been used to obtain homogeneously distributed samples8,9,10,11. The former method requires capping agents that may severely limit the catalytic activity. Therefore, the use of high-surface-area materials as supports is a promising strategy for the preparation of ultrafine and well-defined noble-metal NPs that not only function as barriers to prevent encapsulated NPs from coalescing but also improve the chemical and thermal stability and enhance the electrical conductivity of functional materials. Various noble metal based catalytic systems have been developed, such as Au@ZrO2, Au@Carbon and Pt-decorated carbon composites12,13,14. For example, the Au@Carbon core-shell catalyst exhibits excellent catalytic properties and it can catalyze the reduction of 4-nitrophenol (4-NP) by NaBH4 to 4-aminophenol (4-AP) within 5 min at room temperature13. The facile and green synthesis of novel metal-NP catalysts that exhibit further improved catalytic activity remains a challenge.
In this work, we fabricated a high-performance Pd–rGO–CNT nanocomposite catalyst by depositing Pd NPs onto a reduced graphene oxide (rGO)–carbon nanotube (CNT) support. We synthesized a three-dimensional (3D) porous rGO–CNT hydrogel by hydrothermally treating an aqueous dispersion of graphene oxide (GO) and CNTs. We obtained the Pd–rGO–CNT composites by dipping the rGO–CNT composites into aqueous solutions of K2PdCl4 for a few minutes. Pd NPs with a uniform size of 4 nm were grown in situ on GO–CNT scaffolds by the spontaneous redox reaction between a noble-metal precursor and carbon materials. The ideal combination of Pd NPs and the 3D rGO–CNT framework provides the resultant Pd–rGO–CNT hybrid with integrated properties, such as large surface area, high loading of catalytic NPs and good stability and durability. Therefore, the as-prepared nanohybrid exhibited remarkably high and stable catalytic activity toward the reduction of 4-NP. The reduction of 4-NP by NaBH4 to 4-AP was completed within 20 s at room temperature. The catalyst was easily separated from the product and was reused directly. As far as we know, this material is the most effective nanohybrid catalyst for 4-NP reduction. Our method offers several advantages in terms of easy scale-up, environmentally friendliness and low cost. We envision that the broad application of this strategy will contribute to the development of the next generation of advanced catalytic materials.
Results
The synthetic route for the Pd–rGO–CNT composite is shown in Fig. 1. GO prepared via the Hummer method contains a large number of oxygen-containing functional groups, which allow GO to be well-dispersed in an aqueous solutions15. The amphiphilic nature of GO sheets can serve as surfactant; CNTs can be dispersed into individual ones after ultrasonication and adsorbed onto the GO surface through π–π attractions16. Hydrothermal treatment of the GO and CNT aqueous dispersion leads to the formation of macroscopic rGO–CNT cylinder hydrogels via weak interactions, such as van der Waals forces, hydrogen bonding, π–π stacking and inclusion interactions (Fig. S1). These hydrogels have well-defined and interconnected three-dimensional (3D) porous network and the pore walls consist of thin layers of stacked rGO sheets (Fig. 2a). The partial coalescing or overlapping of graphene sheets results in the formation of cross-linking sites of the framework of hybrid and the pore sizes of the network are in the range of several submicrometer to micrometers. The CNTs are uniformly distributed between the graphene sheets. The surface area of the 3D rGO–CNT material was 307 m2g−1, which is greater than that of the 3D rGO material (201 m2g−1) obtained by fitting the isotherm to the Brunauer–Emmett–Teller (BET) model (Fig. S2). This increased surface area is ascribed to the synergistic effects between the graphene and CNTs in the rGO–CNT nanocomposite. During the self-assembly process of rGO–CNT hydrogels, rGO easily forms agglomerates due to the π–π interactions between individual graphene nanosheets. The insertion of hierarchical tortuous CNTs in rGO–CNT nanocomposites can bridge the adjacent graphene nanosheets, prevent graphene sheets from restacking and increase the specific surface area.

Schematic illustration of the synthesis of the Pd–rGO–CNT composite.

SEM and TEM images of Pd–rGO–CNT composite.
(a) SEM image of Pd–rGO–CNT hybrid. (b–d) TEM images of Pd–rGO–CNT hybrid. (e) The selected-area electron diffraction (SAED) pattern and (f) XRD patterns of Pd–rGO–CNT composite.
The Pd–rGO–CNT composite was obtained via a redox reaction between the rGO–CNT composites and a K2PdCl4 aqueous solution in an ice bath. During this process, the 3D-structured rGO–CNT hydrogel not only increases the specific contact area, which facilitates transport of the Pd2+ ions and electrons into the inner region of the graphene sheets, but also provides more exposed active sites for the anchoring of Pd precursors and for Pd nucleation. Transmission electron microscopy (TEM) images revealed that Pd NPs with a uniform size of 4 nm were well-dispersed on the surfaces of the rGO–CNT scaffolds (Figs. 2b–d). The corresponding particle size distribution histogram of Pd nanoparticles is shown in Fig. S3. The interplanar spacings for the lattice fringes of Pd were 0.223 nm and 0.195 nm, which correspond to the (111) and (200) lattice planes, respectively, of the face-centered cubic (fcc) Pd structure (Fig. 2d). The selected-area electron diffraction (SAED) pattern corresponds to the (111), (200), (220) and (311) planes of the expected fcc Pd (Fig. 2e). XRD patterns and the corresponding standard line of Pd are shown in Fig. 2f. The strongest reflection of the rGO–CNT composite is the (002) reflection at 2θ = 25.9°, which also proves that the Pd NPs exhibit an fcc pattern typical of Pd metal. In addition, X-ray energy dispersive spectroscopy (EDS) spectra further confirm the presence of metallic Pd (Fig. S4).
Quantitative information related to the chemical state, type and extent of the surfaces of the GO–CNT, rGO–CNT and Pd–rGO–CNT are investigated by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 3. Typically, the peaks at 284.6, 286.6 and 288.2 eV are assigned to the C−C, C−O and C = O bonds, respectively. The peak intensity of the C−O bond in GO was the most intense (54.57%), due to the presence of oxygen-containing functional groups (Figs. 3b–d). After the hydrothermal reduction process, the peak intensity of the C–O bond decreased to 17.72% (Fig. 3c). Furthermore, after the sample was immersed in the K2PdCl4 solution, the peak intensity of the C–O bond increased from 17.72% to 21.96% and peaks corresponding to the binding energies of the binding energies of Pd 3d5/2 and 3d3/2 (335.2 and 340.2 eV, respectively) appeared (Fig. S5). Quantitative analysis of the XPS spectrum provided compelling evidence that the Pd content in the Pd–rGO–CNT composite is 1.33 wt%, as shown in Supporting Information (Table S1). In addition, the Pd content was determined using microwave plasma-atomic emission spectrometer (MP-AES). The results showed that the Pd concentration in Pd–rGO–CNT is 1.12 wt%, which is approximately consistent with the concentration obtained from the XPS spectrum. These results suggest that the nucleation of a Pd particle on rGO–CNT is attributed to the direct redox reaction between the metal ions and the rGO–CNT composite. According to the previous studies, the Fermi Level, which is equivalent to the electrochemical potential of electrons in a redox system of CNT, is approximately about +0.5 V vs SHE17,18,19 and the reduction potential of rGO is less than 0.25 V vs SHE20,21, both of these potentials are lower than the reduction potential of PdCl42− (+0.6 V vs SHE). Therefore, rGO/Pd2+ and CNT/Pd2+ systems undergo spontaneous oxidation and reduction in solution.

(a) X-ray photoemission spectroscopy (XPS) profiles of GO–CNT, rGO–CNT and Pd–rGO–CNT hybrid samples. (b) Curve fit of the C1s peak of GO–CNT. (c) Curve fit of the C1s peak of rGO–CNT. (d) Curve fit of C1s peak of Pd–rGO–CNT.
We evaluated the catalytic ability of the as-synthesized Pd–rGO–CNTs in the reduction of 4-NP to 4-AP by NaBH4 in an aqueous medium22,23. The addition of Pd–rGO–CNT caused fading and ultimately bleaching of the yellow color of the 4-NP in aqueous solution. The absorption of 4-NP at 400 nm decreased rapidly, with a concomitant increase in the intensity of a new peak at 300 nm in UV-Vis spectra; this peak was assigned to 4-AP (Fig. 4). Whereas reactions in the presence of Au/Carbon core–shell and Au/SiO2 microspheres as catalysts were completed in 300 s and 1 h, respectively, the reduction with the Pd–rGO–CNT composite as catalyst could be performed in only 20 s, even though the Pd content of the catalyst was as low as 1.12 wt%. We investigated the stability of Pd–rGO–CNT by performing the same reduction reaction with the same catalyst 20 times. This catalyst also exhibited high activity after 20 successive cycles of reactions, with 100% conversion within a reaction period of 40 s, which exceeds the performance of the Au/carbon core–shell and the Au/SiO2 systems. In the presence of the Au/carbon core–shell and the Au/SiO2 microsphere catalysts, the reduction reactions were completed in 300 s and 1 h, respectively (Table 1). We evaluated the stability of the Au@carbon catalyst by performing the same reduction reaction five times. The conversion was close to 100% within a reaction period of 5 min.

(a) UV-Vis spectra of 4-NP reduction (i) before and (ii) 20 s after the addition of the Pd–rGO–CNT composite. (b) The stability of the catalyst during 20 cycles of the same reduction reaction.
Such excellent catalytic performance of Pd–rGO–CNT nanocomposite is related to its 3D macrostructures. During hydrothermal treatment, the conjugated structure of the rGO sheets and the CNTs provide stacking sites on which 3D macrostructures are formed. This architecture with its unique conductive carbon networks is an effective scaffold for the deposition of Pd NPs Well-defined Pd NPs that are highly dispersible and have an ultrasmall size can grow in situ on a 3D rGO–CNT framework through the direct redox reaction between metal ions and the rGO–CNT composite, which is the most important issue for improving the catalytic performance and realizing the application of Pd–rGO–CNT nanomaterials.
Discussion
To demonstrate the advantages of the Pd–rGO–CNT material, macroscopic rGO, rGO–CNT and Pd–rGO composites with porous structures were also synthesized for comparison (Fig. S6). Whereas pristine carbon materials, including rGO and rGO–CNT, exhibited no catalytic properties, the Pd–rGO nanocomposite demonstrated lower catalytic activity and stability than the Pd–rGO–CNT nanocomposite. The reaction time increased from 30 s to 210 s after five successive cycles of reactions when Pd–rGO nanocomposite was used as the catalyst. Therefore, the introduction of CNTs is thought to effectively improve the electron-transfer rate24,25, to provide sufficient space and sites for the redox reaction of 4-NP to 4-AP by NaBH4 and to accelerate the reduction rate. At the same time, the microstructure of the rGO–CNT architecture prevents the aggregation of Pd NPs. The resultant Pd–rGO–CNT nanocomposite possesses a large surface area, high mechanical strength and excellent durability; it consequently exhibits remarkably high, stable catalytic activity.
In summary, we developed a facile and green method for the synthesis of 3D Pd–rGO–CNT nanocomposites and explored their practical application in catalysis. This new type of catalyst was synthesized through hydrothermal treatment of an aqueous dispersion of GO and CNT, followed by spontaneous redox reaction between Pd2+ and an rGO–CNT hybrid to form Pd NPs decorated with rGO–CNT microporous materials. The resultant Pd–rGO–CNT nanocomposites possess a unique large surface area, an extremely high aspect ratio, high mechanical strength and excellent durability and therefore exhibit remarkably high and stable catalytic activity toward the reduction of 4-NP. These features, coupled with the simplicity of the assembly process, the suitability of the method for large-scale production and the green synthesis approach, allow a variety of industrial applications in environmental protection, electrocatalysis, biomedical applications and sensors.
Methods
Preparation of GO
GO was synthesized by the modified Hummers method15. Briefly, 10 g of natural graphite and 5 g of NaNO3 were added to 230 ml of concentrated sulfuric acid under stirring in a flask immersed in an ice-water bath. Then, 30 g of KMnO4 was added slowly and the mixture was stirred at 30°C for 2 h. Next, 460 ml of distilled water was added and the mixture was further stirred for 30 min at 95°C. Finally, 940 ml of distilled water and 30 ml of H2O2 (5%) were subsequently added to terminate the reaction and the color of the solution turned from dark-brown to yellow. The generated solid graphite oxide was separated by centrifugation, washed and dried under vacuum.
Self assembly of rGO-CNT hydrogel
The material was prepared by the addition of 30 mg of CNTs to a 2 mg/ml homogeneous GO aqueous dispersion (the mass ratio of GO to CNT was 2:1) under sonication for approximately 30 min. The mixture was subsequently sealed in a 50-ml Teflon-lined autoclave and maintained at 180°C for 12 h. After the mixture was cooled in room-temperature air with natural convection, a black gel-like 3D cylinder was obtained. The size of the hydrogel could be freely adjusted by changing the volume of the GO aqueous dispersion. The as-obtained samples were freeze-dried overnight for use in subsequent experiments. The 3D rGO hydrogel was fabricated by the same method but without the addition of CNTs.
Synthesis of Pd-rGO-CNT nanocomposite
One milliliter of a K2PdCl4 (10 mM) aqueous solution and 5 mg of rGO–CNT composite were added to a 10-ml aqueous solution and maintained in a vial under vigorous stirring for 30 min in an ice bath. Afterwards, the reaction mixture was washed three times with pure water and centrifuged to remove the remaining reagents. The Pd–rGO nanocomposite was synthesized using a similar procedure.
Catalytic study
4-Nitrophenol (3 ml, 0.1 mM) was mixed with a freshly prepared aqueous solution of NaBH4 (0.1 ml, 0.3 M). Different catalysts (5 mg) were then added under constant magnetic stirring. UV-Vis absorption spectra were recorded to monitor the change in the reaction mixture.
Chemicals and apparatus
Natural graphite powder was purchased from Alfa Aesar (USA); multi-walled carbon nanotubes (CNTs) were purchased from XF NANO (China); K2PdCl4 (99%), 4-nitrophenol (>99%) and NaBH4 (96%) were purchased from Sinopharm Chemical Reagent Co. (China) and were used directly without further purification. All other chemicals used were of analytical reagent grade. Ultra-pure water (18 MΩ·cm) for solution preparation was produced using an Aquapro System (China). Scanning electron microscopy (SEM) images were obtained on an FESEM instrument (GSM6510LV, JEOL, Japan). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained using a TECNAI G2-20 U-Twin instrument (Netherlands) operated at an acceleration voltage of 200 kV. The samples of composites were suspended in ethanol and were prepared by being drop-cast onto a carbon-coated 200-mesh copper grid and subsequently dried at room temperature. X-ray photoelectron spectroscopy (XPS) measurements were performed on a VG ESCALAB 250 spectrometer (UK) using an Al Kα X-ray source (1486 eV), X-ray radiation (15 kV and 10 mA) and hemispherical electron energy analyzer. All of the binding energies were calibrated according to the reference energy of C1s (C1s = 284.6 eV). X-ray powder diffraction (XRD) measurements were performed on a diffractometer (X′ Pert PRO, Panalytical B.V., Netherlands) equipped with a Cu Kα radiation source. The Pd content was determined using a microwave plasma-atom emission spectrometer (MP-AES, Agilent 4100, USA). UV-Vis measurements were conducted on a Specord 50 UV-Vis spectrophotometer (Germany). Nitrogen adsorption/desorption isotherms were obtained at 77 K on an accelerated surface area and porosimetry system (ASAP 2020, USA) to measure the surface area of the films using the Brunauer–Emmett–Teller (BET) method.
References
Balanta, A., Godard, C. & Claver, C. Pd nanoparticles for c–c coupling reactions. Chem. Soc. Rev. 40, 4973–4985 (2011).
Bianchini, C. & Shen, P. K. Palladium-based electrocatalysts for alcohol oxidation in half cells and in direct alcohol fuel cells. Chem. Rev. 109, 4183–4206 (2009).
Nishihata, Y. et al. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature. 418, 164–167 (2002).
Li, P., Wu, Z., Peng, Q. & Li, Y. D. Au-ZnO hybrid nanopyramids and their photocatalytic properties. J. Am. Chem. Soc. 133, 5660–5663 (2011).
Chen, S., Wei, Z. D., Qi, X. Q. & Dong, L. C. Nanostructured polyaniline-decorated Pt/C@PANI core–shell catalyst with enhanced durability and activity. J. Am. Chem. Soc. 134, 13252–13255 (2012).
Yin, H. J., Tang, H. J., Wang, D. & Gao, Y. Facile synthesis of surfactant-free Au cluster/graphene hybrids for h igh-performance oxygen reduction reaction. ACS Nano. 6, 8288–8297 (2012).
Li, Y. J. et al. Stabilization of high-performance oxygen reduction reaction Pt electrocatalyst supported on reduced graphene oxide/carbon black composite. J. Am. Chem. Soc. 134, 12326–12329 (2012).
Pileni, M. P. The role of soft colloidal templates in controlling the size and shape of inorganic nanocrystals. Nat. Mater. 2, 145–150 (2003).
Wilson, O. M., Scott, R. W. J., Garcia-Martines, J. C. & Crooks, R. M. Synthesis, characterization and structure-selective extraction of 1-3-nm diameter AuAg dendrimer-encapsulated bimetallic nanoparticles. J. Am. Chem. Soc. 127, 1015–1024 (2005).
Son, S. U. et al. Facile synthesis of various phosphine-stabilized monodisperse palladium nanoparticles through the understanding of coordination chemistry of the nanoparticles. Nano. Lett. 4, 1147–1151 (2004).
Lee, Y., Garcia, M. A., Frey Huls, N. A. & Sun, S. H. Synthetic tuning of the catalytic properties of Au-Fe3O4 nanoparticles. Angew. Chem. Int. Ed. 49, 1293–1296 (2010).
Arnal, P. M., Comotti, M. & Schiith, F. High-temperature-stable catalysts by hollow sphere encapsulation. Angew. Chem. Int. Ed. 45, 8404–8407 (2006).
Liu, R. et al. Dopamine as a carbon source: The controlled synthesis of Hollow carbon spheres and yolk-structured carbon nanocomposites. Angew. Chem. Int. Ed. 50, 6799–6802 (2011).
Ikeda, S. et al. Ligand-free platinum nanoparticles encapsulated in a hollow porous carbon shell as a highly active heterogeneous hydrogenation catalyst. Angew. Chem. Int. Ed. 45, 7221–7224 (2006).
Hummers, W. S. & Offeman, R. E. Preparation of graphitic. oxide. J. Am. Chem. Soc. 80, 1339 (1958).
Zhou, Y., Bao, Q. L., Tang, L. A., Zhong, Y. L. & Loh, K. P. Hydrothermal dehydration for the “green” reduction of exfoliated graphene oxide to graphene and demonstration of tunable optical limiting properties. Chem. Mater. 21, 2950–2956 (2009).
Suzuki, S., Bower, C., Watanabe, Y. & Zhou, O. Work functions and valence band states of pristine and Cs-intercalated single-walled carbon nanotube bundles. Appl. Phys. Lett. 76, 4007–4009 (2000).
Kazaoui, S., Minami, N., Matsuda, N. H, H. K. & Achiba, Y. Electrochemical tuning of electronic states in single-wall carbon nanotubes studied by in situ absorption spectroscopy and ac resistance. Appl. Phys. Lett. 78, 3433–3435 (2001).
Memmingm, R. Photoinduced Charge Transfer processes at semiconductor electrodes and particles. Top. Curr. Chem. 169, 105–107 (1994).
Choi, H. C., Shim, M., Bangsaruntip, S. & Dai, H. J. Spontaneous reduction of metal Ions on the sidewalls of carbon nanotubes. J. Am. Chem. Soc. 124, 9058–9059 (2002).
Chen, X. M. et al. Synthesis of “Clean” and Well-Dispersive Pd nanoparticles with excellent electrocatalytic property on graphene oxide. J. Am. Chem. Soc. 133, 3693–3695 (2011).
Panigrahi, S. et al. Synthesis and size-selective catalysis by supported gold nanoparticles: study on heterogeneous and homogeneous catalytic process. J. Phys. Chem. C. 111, 4596–4605 (2007).
Lee, J., Park, J. C. & Song, H. A Nanoreactor framework of a Au@SiO2 yolk/shell structure for catalytic reduction of p –nitrophenol. Adv. Mater. 20, 1523–1528 (2008).
Kim, J. et al. Graphene oxide sheets at Interfaces. J. Am. Chem. Soc. 132, 8180–8186 (2010).
Tung, V. C. et al. Surfactant-free water–processable photoconductive all-carbon composite. J. Am. Chem. Soc. 133, 4940–4947 (2011).
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (Project No. 51173055).
Author information
Authors and Affiliations
Contributions
T.S., Z.Z. and J.X. contributed equally. T.S., F.X., S.W. and Y.L. proposed, planned and designed the project. T.S., Y.Z. and C.C. performed the material preparation, characterizations and catalytic tests. All authors contributed to writing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information-Facile and Green Synthesis of Palladium Nanoparticles-Graphene-Carbon Nanotube Porous Material with High Catalytic Activity
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Sun, T., Zhang, Z., Xiao, J. et al. Facile and Green Synthesis of Palladium Nanoparticles-Graphene-Carbon Nanotube Material with High Catalytic Activity. Sci Rep 3, 2527 (2013). https://doi.org/10.1038/srep02527
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02527
This article is cited by
-
Calix[4]pyrrole Stabilized PdNPs as an Efficient Heterogeneous Catalyst for Enhanced Degradation of Water-Soluble Carcinogenic Azo Dyes
Catalysis Letters (2021)
-
Immobilizing biogenically synthesized palladium nanoparticles on cellulose support as a green and sustainable dip catalyst for cross-coupling reaction
Cellulose (2020)
-
Simultaneous growth of three-dimensional carbon nanotubes and ultrathin graphite networks on copper
Scientific Reports (2019)
-
Electroconductive performance of polypyrrole/reduced graphene oxide/carbon nanotube composites synthesized via in situ oxidative polymerization
Journal of Materials Science (2019)
-
Electrochemical properties of Na0.5Bi0.5TiO3 perovskite as an anode material for sodium ion batteries
Journal of Materials Science (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
