
Abstract
Development of tolerance to endotoxin prevents sustained hyper inflammation during systemic infections. Here we report for the first time that chronic morphine treatment tempers endotoxin tolerance resulting in persistent inflammation, septicemia and septic shock. Morphine was found to down-regulate endotoxin/LPS induced miR-146a and 155 in macrophages. However, only miR-146a over expression, but not miR-155 abrogates morphine mediated hyper-inflammation. Conversely, antagonizing miR-146a (but not miR-155) heightened the severity of morphine-mediated hyper-inflammation. These results suggest that miR-146a acts as a molecular switch controlling hyper-inflammation in clinical and/or recreational use of morphine.
Similar content being viewed by others

Introduction
Morphine is the most preferred analgesic used by surgeons in post-operative pain management and also a prevalent drug of abuse1,2,3,4. In either circumstances, the adverse effects of chronic morphine on the immune system has been well documented over the years (for a recent review, see Roy 20113). Higher levels of morphine in systemic circulation reduces pathogen clearance in rodents and human patients, specifically in case of opportunistic infection2,3,4,5 and also induces translocation of gut microbes into the peritoneal organs including mesenteric lymph nodes (MLN), liver, kidneys and eventually, the blood stream6,7. Recent studies have shown that morphine and μ-opioid receptor (MOR) mediated signaling mechanisms are compromised in oral mucosa, leading to severe Salmonella loads8,9. Initially, morphine mediated translocation of gut commensal bacteria was not implicated in persistent inflammation or sepsis10, however, several subsequent studies have reported gut-derived lipopolysaccharide (LPS) mediated inflammation, leading to sepsis and septic shock in rodents undergoing chronic morphine treatment1,11,12,13. In the context of an organism’s response to an inflammatory stimulus, morphine is known to disrupt the tightly balanced natural tolerance to inflammation11,13. Conversely, endogenous morphine levels are typically higher in patients with acute sepsis and septic shock, compared to healthy controls14.
LPS is a gram-negative bacteria-derived putative endotoxin15, which activates the complement system and induces the immune cells, e.g. macrophages and neutrophils to secrete pro-inflammatory cytokines like tumor necrosis factor- alpha (TNF-α) and interleukin-1 (IL-1)13,16, interleukin (IL)-6 and prostaglandins17, thereby inducing persistent inflammation and state of sepsis. Lack of efficient bacterial clearance, as evident in chronic morphine regimen, continued translocation and microbial proliferation result in increasing amount of endotoxin and leads to a state of septic shock13,18. LPS is a cognate ligand for toll-like receptor (TLR) 4, which is a part of the germline-encoded pattern recognition receptor (PRR) family, specially empowered to recognize the Pathogen-Associated Molecular Patterns (PAMPs) of endogenous and/or invading microbes19. TLR4 forms a heterodimer with the myeloid differentiation factor 2 (MD2), resulting in a functional LPS recognition receptor20 and upon stimulation, recruits the myeloid differentiation primary response gene (MyD)88, which in turn recruits the IL-1R-associated kinases (IRAK)-4. Activated IRAK-4 further activates IRAK-1 and 2 and interacts with the E3 ubiquitin ligase TNF receptor-associated factor (TRAF)-6, which ubiquitinylates a complex of TGFβ-activated kinase 1(TAK1), TAK1-binding proteins (TAB)-1,2 and 3, thereby mediating the degradation of the nuclear factor-kappa B (NF-κB) inhibitor IκB. Activated NF-κB migrates to the nucleus and upregulates the pro-inflammatory cytokines19,21. Interestingly, in healthy individuals, both sepsis and consequent exposure to LPS is characterized by an initial hyper-production of cytokines, followed by a “silencing” phase, where TLR-mediated production of pro-inflammatory cytokines is suppressed. This has been variously referred to as “TLR reprogramming” or “endotoxin/LPS tolerance”22. The mechanisms proposed for the endotoxin tolerance range from silencing of key mediators of TLR signaling23 to impaired interaction between different signaling mediators and overexpression of A20, a key de-ubiquitinylation enzyme22. Within the past decade, while the “silencing” mechanism is increasingly being implicated in describing drug/endotoxin tolerance, a new class of molecules, namely the micro-RNAs (miRNA) have emerged as key players in selectively silencing the intermediaries of TLR signaling between the surface receptor and eventual NF-κB activation24,25,26,27,28,29,30,31,32,33,34,35,36.
MicroRNAs are a conserved class of endogenous non-coding RNAs of approximately 22 nucleotides, which modulate the post-transcriptional expression of specific genes by controlling the stability and/or translation of target mRNAs26,37. In mammals, miRNAs have since been implicated in various cellular and systemic functions, like cell differentiation38, cancer39,40, viral infection41 and insulin regulation42. In terms of endotoxin tolerance, various miRNAs (miR-146a, miR-132 and miR-155) are shown to be upregulated in response to LPS and target TAB2, IRAK1 and TRAF6, thereby preventing NF-κB activation27,31,32,34. Recently, miR-221, miR-579 and miR-125b have been shown to be upregulated in response to LPS and target TNFα expression by way of transcriptional silencing and translational disruption28,29, thus establishing a significant role of miRNAs in endotoxin tolerance.
In this study, we investigated the role of miRNAs in modulating LPS tolerance in the context of chronic morphine use (clinical or abuse), leading to severe inflammation and septicemia. Our initial studies showed morphine-mediated dampening of the hyper-inflammation typically associated with early sepsis, followed by a persistent hyper-inflammation compared to the tolerized placebo/saline controls in vitro and in vivo. Subsequent miRNA microarray studies revealed significant inhibition of LPS induced miR-146a and miR-155 levels in murine macrophages undergoing chronic morphine treatment, which was further validated individually by qPCR. We also saw a significant downregulation of IRAK-1 and TRAF-6 (target for miR-146a) and TAB-2 (target for miR-155) over time upon LPS treatment, an effect, not seen in presence of chronic morphine. Next, we show that over-expression of the microRNA inhibitors of the two miRNA results in the loss of LPS tolerance in placebo treated mice and exacerbate the inflammatory response in the morphine treated mice leading to severe septicemia and septic shock, where miR-146a (not miR-155) seems to play a major role. Finally, we demonstrate a complete loss of LPS induced inflammation and morphine mediated attenuation of tolerance in mice overexpressing the two microRNAs, implying for the first time that chronic morphine mediated sepsis and loss of endotoxin tolerance is tightly regulated by a micro-RNA mechanism.
Results
Morphine abrogates LPS tolerance in vivo and in vitro
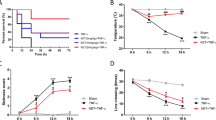
Continuous presence of LPS and resultant stimulation of TLRs lead to the pro-inflammatory “cytokine storm”. This is followed by the tolerance phenomenon, where the inflammation is seen to be suppressed in a time dependent manner22. To test the effect of chronic morphine on endotoxin tolerance, we measured the systemic levels of IL-6 in placebo or morphine implanted wild-type C57BL/6 (WT) mice every 24 hours as described in methods. As expected, in placebo-implanted mice, we found a significant up-regulation of serum IL-6 at 24 hours and the setting of tolerance by 48 hours ( Fig. 1A ). In mice with chronic morphine however, we found a biphasic effect with an initial inhibition of LPS induced IL-6 levels, followed by significant elevation of the cytokine levels, carried up to the end-point at 72 hours. Morphine, by itself, did not seem to have any significant effect on serum IL-6 levels ( Fig. 1A ). Since monocytes and macrophages have been largely implicated in systemic endotoxin tolerance22,43, we next pre-conditioned the mice with placebo/morphine and LPS for varying periods of time (12 hours-5 days) and splenic macrophages were isolated at the indicated time points. These were re-stimulated ex-vivo with LPS and IL-6 levels were measured from the culture supernatant at indicated time-points ( Fig. 1B ). An initial surge in IL-6 levels, followed by tolerance within 24 hours was observed in placebo/saline treated macrophages, whereas, continuous up-regulation and maintenance of high levels of LPS induced IL-6 levels were seen in animals that were morphine treated. Next, we tested this phenomenon in vitro with the murine macrophage cell line J774.1 and witnessed significant attenuation of LPS tolerance in the morphine treated cells ( Fig. 1C ). Finally, we isolated primary human monocyte-derived macrophages and preconditioned the cells with saline/morphine for 24 hours and 50 ng/ml LPS for the next 6 hours. This was followed by a re-stimulation of the cells with 100 ng/ml LPS for an initial 3 hours and 6 hours thereafter. Between all these steps, the culture supernatant was completely removed and the cells washed with warm phosphate buffered saline (PBS). Here, we observed a drastic onset of LPS tolerance after 9 hours of re-stimulation and its maintenance up to the 15 hour end-point, while the morphine treated macrophages showed significantly higher LPS induced IL-6 levels compared to the saline counterparts at the later time points ( Fig. 1D ). These results indicate that chronic maintenance of morphine is able to reverse LPS tolerance in all the models we studied and that monocytes/macrophages seem to have a significant contribution to the phenomenon.

Natural LPS tolerance is abrogated by chronic morphine treatment.
(a) Serum IL-6 levels from mice implanted with placebo or morphine pellets for 24 hours and injected with 1 mg/kg LPS every 24 hours thereafter (n = 5/group; *p < 0.05 compared to PBS24 placebo, **p < 0.05 compared to respective placebo groups, ***p < 0.05 compared to peak placebo IL6 response at 24 hours). (b) Secreted IL-6 levels from cultured splenocytes from mice pre-conditioned with placebo/morphine pellets and LPS for 5 days and re-stimulated with 100 ng/ml LPS ex-vivo for the indicated time points (n = 5/group). (c) Secreted IL-6 levels from murine macrophage cell line J774.1 pre-conditioned with saline/1 μM morphine for 24 hours and treated with 100 ng/ml LPS in continuous presence of saline/morphine. Data representative of at least three independent iterations (*p < 0.05 compared to PBS24 placebo, **p < 0.05 compared to respective placebo groups, ***p < 0.05 compared to peak placebo IL6 response at 24 hours). (d) Secreted IL-6 levels from primary human circulating monocytes/macrophages, pre-conditioned with saline/1 μM morphine for 24 hours and treated with 50 ng/ml LPS for the first 6 hours, followed by 100 ng/ml LPS for subsequent 3 and 6 hours. Data representative of 2 independent healthy donors and two experimental iterations per donor (*p < 0.05 compared to respective placebo groups, ***p < 0.05 compared to peak response at 6 hours).
Morphine modulates LPS induced expression of miR-146a and miR-155
To determine the role of miRs in LPS tolerance and morphine induced persistent activation, WT and mu-opioid receptor knock-out (MORKO) mice, implanted with placebo or morphine pellets were treated with 1 mg/kg LPS for 48 hours and total RNA isolated from peritoneal macrophages was subjected to miRNA array analysis to determine expression profile ( Fig. 2A ). Among the 470 miRNAs analyzed, about 120 of them showed significant up-regulation. MiR-155 and miR-146a showed highest and substantial induction at 48 hr following LPS treatment. Chronic morphine treatment in WT mice significantly decreased LPS-induced mir-155 and mir-146a in the macrophages. Morphine induced inhibition was abolished in the MORKO mice, implying a mu-opioid receptor (MOR) mediated modulation. To further validate the results, WT C57BL/6 and MORKO mice were implanted with placebo or morphine pellets and after 24 hours, injected with 1 mg/kg LPS. The mice were sacrificed and the splenic macrophages were harvested 48 hours post injection, as described in methods and the miR levels analyzed with qPCR. Both miR-155 ( Fig. 2Bi ) and miR-146a ( Fig. 2Bii ) were seen to be significantly up-regulated with LPS and chronic morphine significantly down-regulated the LPS induced expression of the microRNAs, with miR-146a showing more dramatic down-regulation compared to miR-155 ( Fig. 2B ). The fact that this phenomenon is MOR mediated is seen when the MORKO mice were similarly processed and the micro RNA levels measured. For both miRs-146a/155, LPS mediated up-regulation was maintained in presence of chronic morphine in the MORKO mice ( Fig. 2C ). A similar phenomenon was observed in vitro with the J774.1 cell line, where a significant down-regulation of miR-146a was observed within 10 hours of LPS treatment in the context of chronic morphine, which was maintained until 48 hours of treatment ( Fig. 2D ). There was no significant down-regulation of miR-155 beyond 10 hours of LPS treatment ( Fig. 2D ), indicating a greater role of miR-146a compared to miR-155 in LPS tolerance in vitro. In all cases, however, morphine alone did not have any effects on the microRNA levels.

miR-146a and miR-155 are significantly up-regulated by LPS and morphine down-regulated LPS driven microRNA induction.
(a) Heat-map of miR-146a and miR-155 from peritoneal macrophages isolated from wild type and MORKO mice, implanted with placebo/morphine pellets for 24 hours, subsequently injected with 50 μg/kg LPS i.p. for 48 hours and subjected to miR-array analysis (n = 8/group). (b) Independent validation of the microarray data for miR-146a and 155 with qPCR was done with isolated splenocytes from mice, pre-conditioned with placebo/morphine pellet for 24 hours and injected with 1 mg/kg LPS for 48 hours (n = 5/group; *p < 0.05 compared to control, **p < 0.05 compared to LPS alone microRNA levels). (c) MORKO mice were implanted with placebo/morphine pellets for 24 hours and injected with 1 mg/kg LPS for 48 hours. The mice were sacrificed and the spleen harvested. Total mRNA was isolated from the splenocytes using TRIzol reagent (Invitrogen) and cDNA prepared using the miR-script RT kit (Promega). Real-time qPCR was performedfor miR-146a and 155 to verify the role of MOR in down-regulation of the LPS induced microRNA in the chronic morphine model. Data shows that for both microRNAs, the fold induction due to LPS is unchanged between placebo and morphine, implying that down-regulation of LPS induced miR-146a and 155 is MOR dependent in the WT mice (Fig. 2B) [n = 5; *p < 0.01 compared to respective ‘No LPS’ placebo controls]. (d) Time dependent miR-146a and 155 level changes in J774.1 cell line, pre-conditioned with saline/1 μM morphine and treated with 100 ng/ml LPS for the indicated time points. Data representative of three independent iterations (*p < 0.05 compared to basal at 0 h, **p < 0.05 compared to the placebo group in the same time point).
LPS and morphine modulate miR-146a/155 targets in the TLR-4 signaling pathway
Since LPS is the cognate ligand of TLR4 and LPS tolerance is a function of TLR4 signaling22, we set out to verify the message and protein level changes, if any, in the miR-146a and 155 targets in the signaling pathway. miR-155 targets TAB2 and miR-146a targets IRAK1 and TRAF6 in the TLR4 pathway27,31,32,34. For message levels, we preconditioned J774.1 with PBS or 1 μM morphine for 24 hours, followed by 24 hours stimulation with 50 ng/ml LPS. The cells were re-stimulated with 100 ng/ml LPS for the indicated time points and the total RNA was isolated and analyzed for the IRAK1 and TAB2 message levels. The message levels of IRAK1 were seen to reduce significantly within 24 hours of LPS treatment in the saline group with consistently high levels in the chronic morphine treated groups in all later time points ( Fig. 3Ai ). For TAB2, significant reduction of message levels, consistent with LPS tolerance was witnessed within 24 hours. However morphine treatment did not significantly alter LPS induced TAB2 message levels and levels were comparable to placebo treated animals ( Fig. 3Aii ). For protein levels, J774.1 cells were subjected to treatments as above and western blot analysis was performed with the cell lysate at indicated time points ( Fig. 3B ). Similar to the message levels, IRAK1 protein levels were seen to diminish at all later time points following LPS treatment in the control group. In the morphine treated group, IRAK1 protein levels were not decreased and remained constant at all times tested following LPS treatment. ( Fig. 3B ). Next, we validated this finding in vivo in mice, where WT C57BL/6 mice were implanted with placebo or morphine pellets and treated with 1 mg/kg LPS for 24 hours and 48 hours. Groups of mice were sacrificed at 24 and 48 hours post LPS injection and peritoneal macrophages harvested, lysed and subjected to western blot analysis for the signaling intermediates. For all the intermediates (IRAK1, TAB2 and TRAF6), the protein levels decrease dramatically upon LPS treatment, however in the morphine treated group basal levels of the proteins are maintained at all time points ( Fig. 3C ). The phenomenon of LPS tolerance is dependent on the engagement of MOR signaling ( Fig. 2C ) and presumably on the TLR signaling as well. To verify that, we used TLR4 knockout (TLR4KO) mice, implanted with placebo/morphine pellet and injected with 1 mg/kg LPS, 24 hours post-implantation. Serum level IL-6 was measured 24 and 48 hours post-injection. For all time points, as expected, IL-6 was maintained at basal levels ( Fig. 3D ), indicating that morphine mediated attenuation of tolerance is a function of MOR and TLR co-stimulation and the balance is maintained by subtle changes in the micro RNA levels. This is further verified by higher IRAK1 levels in TLR4KO peritoneal macrophages, compared to WT basal levels, 48 hours after LPS treatment ( Supplementary Fig. 1 ), indicating that LPS mediated up-regulation of miR-146a is TLR4 dependent.

MiR-146a and 155 target components of TLR4 signaling pathway which is modulated by LPS.
(a) qPCR analysis for IRAK1 (miR-146a target) and TAB2 (miR-155 target) in J774.1 murine macrophage cell line. J774.1 cells were pre-conditioned for 24 hours with saline/1 μM morphine and treated with 50 ng/ml LPS for 24 hours. Cells were restimulated with 100 ng/ml LPS for the indicated time points and analyzed for IRAK1 and TAB2 transcript levels. Data represents three independent iterations (*p < 0.05 compared to 0 h placebo, **p < 0.05 compared to the respective placebo group). (b) Protein level changes for IRAK1 and TAB2 in J774.1 cell line when treated as in ‘a’ above. Figure represents truncated western blot images for simplicity. Whole membrane images are shown in Supplementary figure 4. (c) Western blot analysis of IRAK1, TAB2 and TRAF6 (miR-146a target) protein levels from murine peritoneal macrophages. Mice implanted with placebo/morphine pellet for 24 hours were injected i.p. with 1 mg/kg LPS every 24 hours and sacrificed at the indicated time points. Peritoneal lavage was harvested, macrophages isolated and lysed in SDS-PAGE sample buffer (n = 4 per time point). Figure represents truncated western blot images for simplicity. Whole membrane images are shown in Supplementary figure 5. (d) TLR4KO mice were implanted with placebo/morphine pellets for 24 hours and injected with 1 mg/kg LPS for every 24 hours till 48 hours. Blood was drawn at 0, 24 and 48 hours time points from the facial vein and plasma isolated. IL-6 levels in the plasma was measured using ELISA (eBiosciences). Data shows maintenance of basal levels (- - - -) of blood IL-6 even after 48 hours of LPS stimulation in the TLR4KO mice, indicating that both MOR (Fig. 2C) and TLR4 co-stimulation is absolutely necessary for morphine mediated abrogation of tolerance. [n = 5].
Inhibition/over-expression of miR-146a modulates LPS tolerance and morphine-mediated inflammation, both in vitro and in vivo
To further verify if morphine-associated tempering of LPS tolerance is regulated through microRNA mechanism, we used lentivirus-mediated neutralization and over-expression of miR-146a both in vitro and in vivo. J774.1 cells were transduced with ‘miArrest’ microRNA inhibitor expressing pseudovirus against the microRNA or miR-146a over-expressing pseudovirus and 48 hours post-transduction, treated with 100 ng/ml LPS for 24 and 48 hours. IL-6 levels in the culture supernatant were measured as the readout. Antagonizing miR-146a resulted in complete abrogation of LPS tolerance as evident from the 48 h data point, when compared to the same time point in Fig. 1C ( Fig. 4A ). Over-expression of miR-146a, as expected, significantly reduced IL-6 production compared to the antagonized counterparts and also showed lack of tolerance development ( Fig. 4A ). In both cases, there was no significant difference between saline and morphine groups within the same time point. For in vivo studies, WT C57BL/6 mice were implanted with placebo/morphine pellets and injected (i.p.) with microRNA inhibitor/over-expression pseudo-virus. 48 hours post transduction, LPS was injected and serum IL-6 levels measured at 24 and 48 hours time point. Results show ( Fig. 4B ), that antagonizing miR-146a in vivo resulted in significantly high systemic IL-6, even in the 0 h mice (compared to Fig. 1A ; p < 0.05) and this level was maintained, without any sign of tolerance till the 48 h data point in the placebo group. In the morphine group, however, unlike the in vitro samples, an initial non-significant down-regulation of the cytokine at 0 h and 24 h was followed by a significant elevation at 48 h time point. The miR-146a neutralized/morphine treated mice did not survive beyond 48 hours, whereas the placebo were seen to survive, indicating that neutralization of miR-146a could be responsible for morphine mediated septic shock. Overall, the miR-146a neutralized mice presented a highly morbid demeanor right from the 0 h LPS in these experiments for both treatment groups and more so for morphine group beyond 24 h. Persistent expression of miR-146a microRNA inhibitor seems to maintain IRAK1 at a higher than basal levels even after 48 hours of LPS treatment in the peritoneal macrophages ( Supplementary Fig. 1; ‘146a anti’ ), highlighting the efficacy of the microRNA inhibitor and the probable cause for the morbid behavior and hyper-inflammation in the animals under this treatment. Over-expression of miR-146a, on the other hand, completely abolished any LPS mediated inflammation and production of IL-6 for both placebo and morphine treated mice ( Fig. 4B ). Target level manipulation of IRAK1 by antagonizing or over-expressing miR-146a also reflects the efficacy of lentiviral approach in this study ( Supplementary Fig. 1; ‘146a anti’ and ‘146a OE’ ).

LPS tolerance is dependent upon miR-146a levels and its regulation.
(a) Secreted IL-6 levels from J774.1 cells, pre-conditioned with saline/1 μM morphine and transduced with lentivirus, either expressing miArrest microRNA inhibitor for miR-146a or over-expressing miR-146a, for 48 hours and treated with 100 ng/ml LPS for 24 and 48 hours. Data represents three independent iterations (*p < 0.05). ‘- - -’ indicates the basal expression of IL-6 with pseudovirus alone. (b) Systemic IL-6 levels from WT mice, implanted with placebo/morphine pellet for 24 hours, were injected with lentivirus, either expressing miArrest microRNA inhibitor for miR-146a or over-expressing miR-146a, for 48 hours and injected i.p. with 1 mg/kg LPS every 24 hours. Blood was drawn every 24 hours through facial vein and plasma IL-6 levels measured with ELISA (n = 6 per group; *p < 0.05, **p < 0.05 compared to ‘anti-miR-146a’ group from the same time-point; ‘- - -’ indicates the basal expression of IL-6 in WT mice as seen in Fig. 1a).
Inhibition and/or over-expression of miR-155 modulates inflammation and tolerance, but not in the chronic morphine regimen
Similar to the miR-146a studies, we characterized miR-155 effects on inflammation and tolerance using lentivirus-mediated neutralization and over-expression of the microRNA both in vitro and in vivo. J774.1 cells were transduced with pseudovirus containing miR-155 microRNA inhibitor or over-expression cassette for 48 hours and saline/morphine for 24 hours before the addition of 100 ng/ml LPS for 24 and 48 hours thereafter. Results show ( Fig. 5A ) that antagonizing the microRNA still results in some measure of tolerance at 48 h time point, compared to the 24 h, as we see a significant reduction in IL-6 levels at the later time point. The morphine reversal of tolerance, however, was completely abolished and the IL-6 levels were maintained similar to the saline group. Upon over-expressing miR-155, IL-6 levels are seen to reduce significantly compared to the antagonized and the native cells in Fig. 1C ( Fig. 5A ). Similarly, in vivo studies were performed where WT mice were injected with microRNA inhibitor/over-expression lentivirus i.p. for 48 hours and implanted with placebo/morphine pellets for 24 hours, followed by 1 mg/kg LPS injection. Systemic levels of IL-6 were measured from the serum at 24 and 48 hours time points. Unlike in vitro results, antagonizing miR-155 did abolish systemic tolerance in the placebo-implanted mice ( Fig. 5B ). In contrast, the morphine-implanted mice showed significantly lower levels of IL-6 for all time points measured; a clear departure from the miR-146a antagonized, morphine-implanted mice, where we witnessed onset of hyper-inflammation at around 48 h of LPS treatment. Additionally, like the miR-146a antagonized mice, the miR-155 antagonized placebo-implanted animals exhibited morbid behavior from 0 h LPS time point and progressively so upon LPS treatment, albeit with 100% survival till the end of the experiment. Over-expression of miR-155, on the other hand, significantly suppressed systemic IL-6 production with or without LPS stimulation ( Fig. 5B ). To compare between our method of lentivirus-mediated down-regulation of miR-155 and the commercially available miR-155−/− (miR-155 knockout) mice, we implanted the latter with placebo/morphine and injected with 1 mg/kg LPS and serum levels of IL-6 measured after 24 and 48 h. The results show above-basal levels of IL-6 for placebo and morphine implanted mice; similar to the microRNA inhibitor injected mice in Fig. 5B ( Fig. 5C ).

LPS tolerance is dependent upon miR-155 levels but not in the chronic morphine regimen.
(a) Secreted IL-6 levels from J774.1 cells, pre-conditioned with saline/1 μM morphine and transduced with lentivirus, either expressing miArrest microRNA inhibitor for miR-155 or over-expressing miR-155, for 48 hours and treated with 100 ng/ml LPS for 24 and 48 hours. Data represents three independent iterations (*p < 0.05). ‘- - -’ indicates the basal expression of IL-6 with control pseudovirus alone. (b) Systemic IL-6 levels from WT mice, implanted with placebo/morphine pellet for 24 hours, were injected with lentivirus, either expressing miArrest microRNA inhibitor for miR-155 or over-expressing miR-155, for 48 hours and injected i.p. with 1 mg/kg LPS every 24 hours. Blood was drawn every 24 hours through facial vein and plasma IL-6 levels measured by ELISA (n = 5 per group; **p < 0.05, ***p < 0.05 compared to ‘anti-miR-155’ group from the same time-point; ‘- - -’ indicates the basal expression of IL-6 in WT mice as seen in Fig. 1a). (c) miR-155 knockout (miR-155 KO) mice were implanted with placebo/morphine pellets for 24 hours and injected with 1 mg/kg LPS for every 24 hours till 48 hours. Blood was drawn at 0, 24 and 48 hours time points from the facial vein and plasma isolated. IL-6 levels in the plasma were measured using ELISA (eBiosciences). Data shows elevated levels of IL-6 in the blood, compared to WT basal (- - - -) and comparable to Figure 5B where we show elevated IL-6 levels in the lentivirus mediated miR-155 neutralized mice. Noticeably, in both cases, LPS tolerance is seen to be obliterated [n = 5].
Discussion
One of the primary responses of the mammalian system, confronted with pathogenic attack, is to up-regulate an acute inflammatory cytokine “storm”, which serves as a “call signal” for the mobilization of the immune mediators22. While this is an important aspect of innate or cell-mediated immunity, the process itself warrants tight regulation to avoid hyper-inflammation owing to self-induced or prolonged activation of inflammation leading to sepsis or septic shock14,20. As understood today, most of this initial response is mediated through the activation of Pathogen Recognition Receptors(PRR)12,15,19,20,21,43,44,45 and the regulation is mediated by modulating the activity or levels of adaptor molecules, downstream of the PRR signaling, leading to endotoxin tolerance28,29,31. Undoubtedly, of all the mechanisms suggested for affecting tolerance, micro RNA mediated silencing of PRR downstream intermediaries, is gaining more ground as evidenced by recent literature22,23,24,25,26,27,28,29,30,31,32,33,34,35,36. Diverse factors, including sepsis, trauma and acute pancreatitis have been implicated in the disruption of the tight regulation of endotoxin tolerance46. Interestingly, clinical or recreational use/abuse of morphine has been shown to be associated with sustained immune activation and promotes sepsis and septic shock. While the systemic analgesic response to morphine undergoes tolerance with chronic use, the effects of morphine on immune cells do not. Adenylate cyclase superactivation, the cellular hallmark of analgesic tolerance contributes to differential activation of signaling pathways in immune cells. Although the physiological relevance for sustaining inflammation in the context of morphine is not entirely understood, several clinical and animal studies show that persistent peripheral inflammation significantly attenuate morphine analgesic tolerance. Thus, there is delay in the development of analgesic tolerance when chronic morphine is administered in the context of pain, suggesting that inflammatory cues sustain morphine’s analgesic response, on the other hand, persistent activation of the mu opioid receptor in the absence of pain cues lead to rapid analgesic tolerance1,2,4,6,8,9,10,11,19,47,48. However the mechanism underlying this morphine-induced reversal of endotoxin tolerance has not been clearly delineated.
In this study, we have demonstrated the attenuation of LPS tolerance both in vitro, in vivo, ex vivo in mice and in vitro in human cells by morphine. We have further shown that both miR-146a and miR-155 are significantly up regulated in response to LPS stimulation and chronic morphine is able to significantly reduce these levels. We show that LPS mediated up-regulation or morphine mediated reversal of the up-regulation of the microRNAs are reflected in the microRNA target levels in the relevant cells and finally, depletion or over-expression of the microRNAs abrogates LPS tolerance, establishing that the microRNAs play a definite role in endotoxin tolerance.
Our microRNA-array experiment highlighted miR-146a and miR-155 as being significantly induced by LPS at 48 hours, consistent with their perceived role in regulating inflammation and tolerance27,31,32,34. In our validation studies, we show LPS driven induction of both the microRNAs and beyond that, a significant departure in the downstream activity of the two, not so much in terms of IL-6 induction, but surely in terms of regulating tolerance. While miR-146a exhibits a strong correlation between LPS induced up-regulation, target ablation in macrophages, resulting in systemic tolerance (or reversal by morphine), miR-155 seems to be inconsistent on all counts except LPS driven induction and macrophage target ablation. As indicated in THP1 cells earlier31,32 and also in our study, unlike miR-155, miR-146a indeed plays a direct role in endotoxin tolerance and especially, morphine’s role in reversal of tolerance in our murine model.
The two microRNAs, in their depletion or over-expression, exhibit differential dynamics in terms of systemic IL-6 production, which could be attributed to differential regulation of their targets in the different cell-types. MiR-155 has been variously implicated in different cell types to promote a pro-inflammatory environment by targeting several anti-inflammatory factors49,50,51. Conversely, anti-inflammatory mediators like glucocorticoids and small molecule drugs have been shown to work through down-regulation of miR-155 in immune cells52,53. The wide plethora of well characterized miR-155 targets, including cytidine diaminase and PU.1 (immunoglobulin class-switching to IgG in B cells), src homology-2 domain containing inositol-5-phosphatase 1 (SHIP1; myeloproliferative disorder)49 and cytotoxic T-lymphocyte antigen-4 (CTLA4; negative regulator of T-cell proliferation)50 are anti-inflammatory in effect, with the sole exception of TAB2. Hence, it is conceivable that upon induction, miR-155 may have pro- or anti-inflammatory effects, depending on the major target being depleted. Indeed, when we compare the SHIP1 versus TAB2 levels in total mice splenocytes, where we also over-express miR-155, we see a complete depletion of SHIP1, while TAB2 levels are nominally reduced after 48 h of LPS treatment ( Supplementary Fig. 2 ). This would explain the above-basal systemic IL-6 seen upon miR-155 over-expression in Fig. 5B . Under similar conditions, in presence of morphine however, we do see an above-basal expression of both the mediators and the level of systemic inflammation would probably depend on the trade-off between pro- and anti-inflammatory effects mediated by the different cell types involved ( Supplementary Fig. 2 ). In our model, we see chronic morphine and miR-155 over-expression depleting IL-6 to below-basal levels ( Fig. 5B ).
A combination of facts, including our previous finding that chronic morphine up-regulates TLR4 expression in glial macrophages54, TLR4’s involvement in opioid reward in mice55 and upregulation of MOR expression in certain situations including tolerance and in the presence of LPS (J774.1 cells) ( Supplementary Fig. 3 ), reveal a multi-layered regulation of innate response to pathogens in the context of chronic morphine regimen. Considering that miR-146a has a major role in this regulation, the scope of morphine-mediated disruption of tolerance may be expanded to a plethora of organisms, since the targets of miR-146a (IRAK1 and TRAF6) participate in all major TLR pathways (except TLR3)32. It may be worthwhile to further dissect the mechanism of tolerance and how morphine disrupts the balance (summarized in Figure 6 ) and consider miR-146a as a molecular switch to control hyper-inflammation in clinical and/or recreational use of morphine.

Model depicting the interplay of miR-146a and miR-155 in different cell types and their contribution to systemic inflammation and LPS tolerance.
 Natural stimulation of TLR4, which on one hand activates NF-κB via a plethora of signaling intermediaries and on the other hand gears up for inducing tolerance and reducing inflammation, by up-regulating miR-146a and 155. Upon activation of the MOR
Natural stimulation of TLR4, which on one hand activates NF-κB via a plethora of signaling intermediaries and on the other hand gears up for inducing tolerance and reducing inflammation, by up-regulating miR-146a and 155. Upon activation of the MOR  , there is a concomitant inhibition of miR-146a and 155, whereby, we see a persistent state of inflammation. In our experiments and in the literature, there is ample evidence that miR-155 has different target preference in different cell types
, there is a concomitant inhibition of miR-146a and 155, whereby, we see a persistent state of inflammation. In our experiments and in the literature, there is ample evidence that miR-155 has different target preference in different cell types  , where down-regulation of SHIP1 or PU.1 promotes inflammation, contrary to the silencing of TAB2 in macrophages. The systemic fallout
, where down-regulation of SHIP1 or PU.1 promotes inflammation, contrary to the silencing of TAB2 in macrophages. The systemic fallout  of this interplay, especially in the context of inflammation is different for miR-146a and 155. While miR-146a seems to tone down TLR4 signaling and NF-κB activation and hence induces tolerance, miR-155 effect is a sum total of individual contribution from different cell types and inflammation would essentially depend on the dominant target of the microRNA under different secondary conditions, including MOR signaling and infection. This clearly implies that miR-146a and not miR-155, would have a direct role to play in a tightly regulated phenomenon like tolerance, specifically in the context of chronic morphine.
of this interplay, especially in the context of inflammation is different for miR-146a and 155. While miR-146a seems to tone down TLR4 signaling and NF-κB activation and hence induces tolerance, miR-155 effect is a sum total of individual contribution from different cell types and inflammation would essentially depend on the dominant target of the microRNA under different secondary conditions, including MOR signaling and infection. This clearly implies that miR-146a and not miR-155, would have a direct role to play in a tightly regulated phenomenon like tolerance, specifically in the context of chronic morphine.
Methods
Mice and cell lines
C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, Maine). TLR4KO and MORKO mice were maintained in-house. All animals were maintained in pathogen-free facilities and all procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee. Typically, 8–10 week old animals were used for our studies. Murine macrophage cell line, J774.1 was obtained from American Type Culture Collection (ATCC; Manassas, VA). Human blood was purchased from The American Red Cross, Minneapolis chapter.
Placebo/morphine pellet Implantation
Slow release morphine pellets (25 mg) and corresponding placebo pellets were kindly provided by National Institute of Drug Abuse (NIDA, National Institutes of Health, Rockville, MD). The implantation procedure involved 3% isoflurane induced anesthesia, followed by making a small incision at the dorsal torso of the mice. The appropriate pellet was inserted into the small pocket created during incision and the wound was closed using stainless steel wound-clips. The whole process was carried out under aseptic conditions.
miR-Array platform
Custom miRNA microarray experiment and analyses were performed as described earlier56. The results have been submitted to the NCBI-GEO database under the accession number GSE42506.
Cell culture and treatments
All cell types used in this study were cultured at 37°C with 5% CO2. For Packaging and production of appropriate pseudo-lentivirus, HEK293TN producer cell-line was purchased from System Biosciences and cultured in DMEM (Gibco), supplemented with 10% FBS and 1% penicillin/streptomycin. These cells were co-transfected with the appropriate miArrest or miExpress plasmids and the packaging plasmid mix using X-tremegene HP transfection reagent (Roche) according to the manufacturer’s specifications. For in vitro studies, J774.1 cells were purchased from ATCC and cultured in DMEM (Gibco), supplemented with 10% FBS and 1% penicillin/streptomycin. For tolerance studies, the cells were preconditioned with 1 μM morphine sulphate (Sigma) or PBS for 24 hours and treated with 50 ng/ml LPS (from E. coli 055:B5; Sigma) for 24 hours, followed by 100 ng/ml for 4, 12, 24, 48 and 72 hours. At each time point, the culture supernatant was saved and replaced with fresh media supplemented with the additives as required. For qPCR studies, J774.1 cells were pre-conditioned with saline/morphine/LPS as above and the cells were harvested at 24 and 48 hours of LPS treatment in 1 ml Trizol reagent (Invitrogen) and processed for RNA isolation, or stored in −80° freezer until further processing. For murine splenic macrophage isolation and ex-vivo analysis, C57BL/6 mice were implanted with placebo or 25 mg slow-release morphine pellet for 24 hours and saline/1 mg/kg LPS for 5 days. The spleens were harvested from these mice, macerated through a 40 μm mesh to obtain a single cell suspension and the erythrocytes lysed using RBC lysis buffer (Sigma). The resultant cells were plated at 37°C with 5% CO2 for 2 hours to adhere the splenic macrophages to the culture dish. At all times, depending on the source, saline/morphine were maintained on these cells ex-vivo. Upon adherence, the cells were re-stimulated with 100 ng/ml LPS for 6,12, 24, 48, 72, 96 and 120 hours and the culture supernatant harvested for cytokine ELISA. Human blood monocyte derived macrophages were similarly allowed to adhere to the culture dish and pre-conditioned with saline/1 μM morphine sulphate for 24 hours and treated with 50 ng/ml LPS for 6 hours. The culture supernatant was replaced with fresh media containing 100 ng/ml LPS for 3 hours and again for another 6 hours. The culture supernatant was saved at −80° freezer for cytokine ELISA.
Lentivirus preparation
miArrest microRNA inhibitor plasmids for miR-146a and 155, the respective over-expression plasmids (miExpress) and the packaging plasmid mix were purchased from Genecopoeia (Rockville, MD). Individual plasmids and the packaging mix were co-transfected into HEK293TN packaging cell-lines using X-tremegene HP transfection reagent (Roche). 48 hours post-transfection, the culture supernatant was harvested and the pseudovirus concentrated using Lenti-pac lentivirus concentration Solution (Genecopoeia). Control plasmid provided with the lentivirus kit was processed as above to obtain the control pseudovirus for secondary transduction of target cells/mice. However, for most experiments, the 0 h time point with the same transduction was used as the control. Multiplicity of Infection (MOI) for each lentiviral preparation was determined using HIV p24 ELISA (Sino Biological) and the standard MOI of 2.0 and above (>85% transduction efficiency) was determined to be approximately 50 transduction units (TU)/pg of p24.
In vitro lentivirus administration
In experiments, where miR-146a or 155 were antagonized or over-expressed, lentivirus was added to the media at calculated concentration of approximately 5 TU/cell, after 24 hours pre-conditioning with saline/1 μM morphine. LPS was added to the media 48 hours post transduction, as described above.
In vivo lentivirus administration
For in vivo injection of lentiviral particles, wherever applicable, approximately 107 TU/mice were injected (i.p.), 24 hours post placebo/morphine pellet implantation. 48 hours post transduction, 1 mg/kg LPS was injected (i.p.) every 24 hours up to 48 hours and in some cases, 72 hours.
In vivo tolerance studies
Mice (C57BL/6, TLR4KO and MORKO, as applicable) were implanted with placebo/morphine pellet and LPS treatment was commenced 24 hours post hence. For studies involving antagonizing or over-expressing miR-146a or 155, lentivirus were injected 24 hours post-implantation and LPS treatment was started 48 hours post-transduction. Every 24 hours, blood samples were drawn from each animal through facial-vein bleeding. At the end point of 48 or 72 hours (as applicable), peritoneal macrophages were harvested by lavage and the spleen was harvested and stored at −80°C till further analysis. Alternatively, cohorts of mice were implanted with placebo/morphine pellets as described and treated with 1 mg/kg LPS (with or without transduction) every 24 hours. Cohorts of mice were sacrificed every 24 hours and peritoneal macrophages (lavage), spleen and blood (direct heart bleeding) were harvested. In all cases, blood were collected into EDTA coated microtainers (BD Biosciences) and the plasma isolated by centrifugation and stored in −80°C until further analysis.
Cytokine assays
IL-6 ELISA kits were purchased from eBiosciences and assay performed according to the manufacturer’s protocol. For each assay, culture supernatant or plasma was quantitated with BCA protein assay kit (Pierce) and IL-6 levels were determined for 1 mg total protein load.
qPCR and primers
Total cellular RNA (J774.1 cells) was extracted using TRIzol (Invitrogen) and cDNA was synthesized with the M-MLV Reverse Transcription Kit (Promega). Primers for IRAK1, TAB2 and 18S ribosomal RNA were purchased from IDT. Primers for OPRM1 (MOR) were purchased from Qiagen. Quantitative real-time polymerase chain reaction (PCR) was performed on an Applied Biosystems 7500 Realtime PCR Detection system. All samples were run in triplicate and relative mRNA expression levels were determined after normalizing all values to 18S RNA. Primer sequence:18s 5’-GTAACCCGTTGAACCCCATT-3’;5’-CCATCCAATCGGTAGTAGCG-3’ IRAK1: 5’-GCTGTGGACACCGATACC-3’;5’-GGTCACTCCAGCCTCTTCAG-3’; TAB2: 5’-ACTACTCCACCGCCAAC-3’; 5’-TTTCTTTGTGGGGGTTCAAG-3’. For miRNA146a and miRNA155, Total cellular RNA was extracted using TRIzol (Invitrogen) and cDNA was synthesized with miScript RT kit (Qiagen). Primers for U6, miR146a and miR155 were purchased from Qiagen. Quantitative real-time polymerase chain reaction (qPCR) was performed on an Applied Biosystems 7500 Realtime PCR Detection system. All samples were run in triplicate and relative mRNA expression levels were determined after normalizing all values to U6 RNA.
Western blot
All antibodies for western blot analysis were purchased from Cell Signaling Technologies except anti-TRAF6 antibody (Sigma). For peritoneal macrophages, the cell pellet was directly dissolved in 1× SDS-PAGE sample buffer (Pierce). For total splenocytes, single cell suspension from spleens were suspended in RBC lysis buffer (Sigma) and RBC-free fraction was directly boiled with the sample buffer as above. Electrophoresis was performed on AnyKd mini gels (Bio-Rad) and transferred onto nitrocellulose membranes (Bio-Rad). The membranes were typically blocked with G-blocker (G Biosciences) overnight and probed with primary antibodies for 2 hours. Membrane was incubated with IRdye (680 and 800; Licor) for one hour, which allowed us to detect the loading control in the same membrane. Signal acquisition was done on Odyssey western blot developer (Licor) as per manufacturer’s instructions.
Statistical analysis
Cytokine concentrations from culture supernatant were expressed as change versus saline ± SD. For plasma level changes, this was expressed as ± SEM. Fold changes in qPCR were expressed as ± SD between groups. Significance was defined as p < 0.05 (ELISA) and p < 0.01 (qPCR) in an unpaired student’s t test.
References
Babrowski, T. et al. Pseudomonas aeruginosa virulence expression is directly activated by morphine and is capable of causing lethal gut-derived sepsis in mice during chronic morphine administration. Ann. Surg. 255, 386–93 (2012).
Roy, S., Wang, J., Kelschenbach, J., Koodie, L. & Martin, J. Modulation of Immune Function by Morphine: Implications for Susceptibility to Infection. J. Neuroimmune. Pharmacol. 1, 77–89 (2006).
Roy, S. et al. Opioid drug abuse and modulation of immune function: consequences in the susceptibility to opportunistic infections. J. Neuroimmune. Pharmacol. 6, 442–65 (2011).
Wang, J., Barke, R. A., Ma, J., Charboneau, R. & Roy, S. Opiate abuse, innate immunity and bacterial infectious diseases. Arch. Immunol. ther. Exp. 56, 299–309 (2008).
Ma, J. et al. Morphine disrupts interleukin-23 (IL-23)/IL-17-mediated pulmonary mucosal host defense against Streptococcus pneumoniae infection. Infec. Immun. 78, 830–7 (2010).
Cuepper, P., Miller, T. & Chen, C. Effective total parenteral nutrition plus morphine on bacterial translocation in rats. J. Parenter. Enteral Nutr. 18, 380–381 (1994).
Ma, L., Deitch, E., Specian, R., Steffen, E. & Berg, R. Translocation of Lactobacillus murinus from the Gastrointestinal Tract. Curr. Microbiol. 20, 177–184 (1990).
MacFarlane, A. S. et al. Morphine increases susceptibility to oral Salmonella typhimurium infection. J. Infect. Dis. 181, 1350–8 (2000).
Breslow, J. M. et al. Potentiating effect of morphine on oral Salmonella enterica serovar Typhimurium infection is μ-opioid receptor-dependent. Microb. Pathog. 49, 330–5 (2010).
Runkel, N. et al. Alterations in rat intestinal transit by morphine promote bacterial translocation. Dig. Dis. Sci. 38, 1530–1536 (1993).
Hilburger, M. E. et al. Morphine induces sepsis in mice. J. Infect. Dis. 176, 183–8 (1997).
Maitra, U. et al. Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J. Immunol. 189, 1014–1023 (2012).
Ocasio, F. M., Jiang, Y., House, S. D. & Chang, S. L. Chronic morphine accelerates the progression of lipopolysaccharide-induced sepsis to septic shock. J. Neuroimmunol. 149, 90–100 (2004).
Glattard, E. et al. Endogenous morphine levels are increased in sepsis: a partial implication of neutrophils. PLoS One. 5, 1–14 (2010).
Lukasiewicz, J. & Lugowski, C. Biologic activity of lipopolysaccharides. Postepy. Hig. Med. Dosw. 57, 33–53 (2003).
Perrillo, J. et al. Septic shock in humans advances in the understanding of pathogenesis, cardiovascular dysfunction and therapy. Ann. Intern. Med. 113, 227–242 (1990).
Itoh, K. et al. Lipopolysaccharide promotes the survival of osteoclasts via toll-like receptor4, but cytokine production of osteoclasts in response to lipopolysaccharide is different from that of macrophages. J. Immunol. 170, 3688–3695 (2003).
Wang, J. H. et al. Bacterial lipoprotein induces endotoxin-independent tolerance to septic shock. J. Immunol. 170, 14–18 (2003).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell. 140, 805–20 (2010).
Park, B. S. et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 458, 1191–5 (2009).
Negishi, H. et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat. Immunol. 13, 659–666 (2012).
Xiong, Y. et al. Endotoxin tolerance impairs IL-1 receptor-associated kinase (IRAK) 4 and TGF-beta-activated kinase 1 activation, K63-linked polyubiquitination and assembly of IRAK1, TNF receptor-associated factor 6 and IkappaB kinase gamma and increases A20 expression. J. Biol. Chem. 286, 7905–16 (2011).
McCall, C. E. & Yoza, B. K. Gene silencing in severe systemic inflammation. Am. J. Respir. Crit. Care Med. 175, 763–7 (2007).
Androulidaki, A. et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity. 31, 220–31 (2009).
Barbalat, R. & Barton, G. M. MicroRNAs and LPS: developing a relationship in the neonatal gut. Cell Host Microbe. 8, 303–4 (2010).
Bartel, D. P., Lee, R. & Feinbaum, R. MicroRNAs: genomics, biogenesis, mechanism and function. Cell. 116, 281–297 (2004).
Cheng, Y. et al. Downregulation of miR-27a* and miR-532-5p and upregulation of miR-146a and miR-155 in LPS-induced RAW264.7 macrophage cells. Inflammation. 35(4), 1308–13 (2012).
El Gazzar, M., Church, A., Liu, T. & McCall, C. E. MicroRNA-146a regulates both transcription silencing and translation disruption of TNF-α during TLR4-induced gene reprogramming. J. Leukoc. Biol. 90, 509–19 (2011).
El Gazzar, M. & McCall, C. E. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J. Biol. Chem. 285, 20940–51 (2010).
Farh, K. K.-H. et al. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 310, 1817–21 (2005).
Nahid, M. A., Pauley, K. M., Satoh, M. & Chan, E. K. L. miR-146a is critical for endotoxin-induced tolerance: implication in innate immunity. J. Biol. Chem. 284, 34590–9 (2009).
Nahid, M. A., Satoh, M. & Chan, E. K. L. Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J. Immunol. 186, 1723–34 (2011).
Ruggiero, T. et al. LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J. 23, 2898–908 (2009).
Taganov, K. D., Boldin, M. P., Chang, K. & Baltimore, D. An inhibitor targeted to signaling proteins of innate immune responses. Proc. Nat. Acad. Sci. 103(33), 12481–12486 (2006).
Thompson, R. C., Herscovitch, M., Zhao, I., Ford, T. J. & Gilmore, T. D. NF-kappaB down-regulates expression of the B-lymphoma marker CD10 through a miR-155/PU.1 pathway. J. Biol. Chem. 286, 1675–82 (2011).
Tili, E. et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179(8), 5082–9 (2007).
Filipowicz, W., Bhattacharyya, S. N. & Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–14 (2008).
Monticelli, S. et al. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 6, R71.1–R71.11 (2005).
He, L. et al. A microRNA polycistron as a potential human oncogene. Nature. 435, 828–33 (2005).
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature. 435, 834–8 (2005).
Lecellier, C.-H. et al. A cellular microRNA mediates antiviral defense in human cells. Science. 308, 557–60 (2005).
Poy, M. N. et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 432, 226–30 (2004).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–84 (2010).
Parrillo, J. Pathogenic mechanism of septic shock. N. Engl. J. Med. 328, 1471–1477 (1993).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell. 124, 783–801 (2006).
Biswas, S. K. & Lopez-Collazo, E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–87 (2009).
Louria, D., Hensle, T. & Rose, J. The major medical complications of heroin addiction. Ann. Intern. Med. 67, 1–22 (1967).
Ninković, J. & Roy, S. Morphine decreases bacterial phagocytosis by inhibiting actin polymerization through cAMP-, Rac-1- and p38 MAPK-dependent mechanisms. Am. J. Pathol. 180, 1068–79 (2012).
O’Connell, R. M. et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 33, 607–19 (2010).
Sonkoly, E. et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J. Allergy Clin. Immunol. 126, 581–589 (2010).
Tili, E. et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–9 (2007).
Tu, J. et al. TanshinoneIIA ameliorates inflammatory microenvironment of colon cancer cells via repression of microRNA-155. Int. Immunopharmacol. 14, 353–361 (2012).
Zheng, Y. et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155, a novel anti-inflammation mechanism. Free Rad. Biol. Med. 52, 1307–17 (2012).
Dutta, R. et al. Morphine modulation of toll-like receptors in microglial cells potentiates neuropathogenesis in a HIV-1 model of coinfection with pneumococcal pneumoniae. J. Neurosci. 32, 9917–30 (2012).
Hutchinson, M. R. et al. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J. Neurosci. 32, 11187–11200 (2012).
Kalscheuer, S., Zhang, X., Zeng, Y. & Upadhyaya, P. Differential expression of microRNAs in early-stage neoplastic transformation in the lungs of F344 rats chronically treated with the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 29, 2394–9 (2008).
Acknowledgements
This work was supported in part by the NIH grants RO1 DA 12104, RO1 DA 022935, RO1 DA031202, K05DA033881, P50 DA 011806 and 1R01DA034582 to SRoy.
Author information
Authors and Affiliations
Contributions
S.B., J.M., S.D., A.K., J.H., R.C. and Y.Z. did experiments. S.B., J.M., S.R. and S. Roy designed experiments. S.B., J.M. and S. Roy wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplimentary Figures
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Banerjee, S., Meng, J., Das, S. et al. Morphine induced exacerbation of sepsis is mediated by tempering endotoxin tolerance through modulation of miR-146a. Sci Rep 3, 1977 (2013). https://doi.org/10.1038/srep01977
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01977
This article is cited by
-
Morphine aggravates inflammatory, behavioral, and hippocampal structural deficits in septic rats
Scientific Reports (2023)
-
Association of miR-155, miR-187 and Inflammatory Cytokines IL-6, IL-10 and TNF-α in Chronic Opium Abusers
Inflammation (2022)
-
Morphine Potentiates Dysbiotic Microbial and Metabolic Shifts in Acute SIV Infection
Journal of Neuroimmune Pharmacology (2019)
-
Myalgic encephalomyelitis or chronic fatigue syndrome: how could the illness develop?
Metabolic Brain Disease (2019)
-
Morphine induces changes in the gut microbiome and metabolome in a morphine dependence model
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
