
Abstract
The 2009 swine-origin H1N1 influenza, though antigenically novel to the population at the time, was antigenically similar to the 1918 H1N1 pandemic influenza and consequently was considered to be “archived” in the swine species before reemerging in humans. Given that the H3N2 is another subtype that currently circulates in the human population and is high on WHO pandemic preparedness list, we assessed the likelihood of reemergence of H3N2 from a non-human host. Using HA sequence features relevant to immune recognition, receptor binding and transmission we have identified several recent H3 strains in avian and swine that present hallmarks of a reemerging virus. IgG polyclonal raised in rabbit with recent seasonal vaccine H3 fail to recognize these swine H3 strains suggesting that existing vaccines may not be effective in protecting against these strains. Vaccine strategies can mitigate risks associated with a potential H3N2 pandemic in humans.
Similar content being viewed by others


Introduction
Influenza A viruses pose a major public health problem, causing seasonal epidemics and occasional—but devastating—global pandemics1 which negatively impact the global economy. Until recently, influenza pandemics were thought to be associated with the introduction of new HA subtypes into the human population2,3,4,5. Indeed, two of the twentieth century pandemics – the 1957–58 H2N2 Asian Flu and the 1967–68 H3N2 Hong Kong Flu - introduced new HA subtypes into the human population4,5. The surface glycoprotein HA of the influenza A virus is the main target of the immune system and mutations on the globular head region (residues 50–230 of HA1, H3 HA numbering used) of this protein determine antigenic novelty, species adaptation and transmission6.
Birds are natural reservoirs for influenza A viruses and avian-adapted viruses either directly crossover to humans (through direct contact) or do so with the help of intermediate swine species. Influenza A viruses rapidly evolve (through antigenic drift) in humans as a consequence of both the complex response of human immune system and rapid geographical movement of human population. In contrast to their rapid antigenic evolution in human hosts, the antigenic evolution of influenza A viruses in avian and swine occurs at a much slower rate7,8,9,10. As a consequence of these factors, the human immunity to past pandemic strains fades over time, thus enabling antigenically “intact” viruses in avian and swine species to reemerge and begin a new infection cycle in humans. For example, although H2N2 subtype does not currently circulate in the human population, viruses carrying HA that are antigenically similar to the 1957–58 pandemic H2N2 virus continue to circulate in avian species11. Among the subtypes that continue to circulate in humans (H1N1 and H3N2), the 2009 H1N1 outbreak offers a practical example of how HA from a swine strain that is antigenically similar to 1918 pandemic H1N1 HA can be reintroduced into the human population12. The question remains of whether this trend is observed in H3N2, given that there has been a high rate of antigenic drift in human H3 subtype13,14,15 since the emergence of 1968 pandemic H3N2. Critically, average hospitalizations and mortality rates were found higher for seasons dominated by A/H3N2 viruses compared to seasons dominated by influenza B or A/H1N116,17,18.
The H3N2 pandemic began in 1968 and was caused by a human-adapted H2N2 virus that obtained avian H3 and PB1 genes through reassortment5. The HA of both 1957 and 1968 pandemic strains are of avian origin. Unlike H2N2, the H3N2 subtype is still in circulation, however the high rate of antigenic drift of human H3 coupled with the long interval since the previous pandemic may mean that the human herd would have ‘forgotten’ the antigenic structure of the 1968 pandemic strain and therefore the reemergence of a similar strain circulating in the avian or swine reservoir could have potentially damaging consequences. Identifying such strains is of paramount value for pandemic surveillance and preparedness.
To address this question in this study we measure the ‘antigenic intactness’ of HA from avian or swine species in reference to HA from the corresponding pandemic subtypes. The antigenic identity (AI) of an avian or a swine HA is defined by the percentage fraction of amino acids in the immunodominant antigenic sites that are conserved in the corresponding pandemic HA (H1, H2 and H3 subtype). The AI value varies between 0 and 100. Values closer to 100 indicate a high antigenic identity with the pandemic HA.
Results
We first applied the AI metric to human-adapted H1N1 and avian H2 subtypes for two reasons. In the former case, we tested the ability of AI values to discriminate the 1918 and 2009 pandemic HAs from the seasonal strains. In the latter case, we validated AI's potential to highlight the conservation of antigenic sites in avian H211. For H1N1, the HA of the human-adapted strains were compared to 1918 pandemic H1N1 HA (A/South Carolina/1/18) and the characterized H1 antigenic sites Sa, Sb, Ca, Cb19,20 were used to calculate AI (Methods). The AI values clearly discriminate the reemerging swine-origin HA of 2009 H1N1 pandemic from the seasonal H1 based on the antigenic identity to the 1918 pandemic H1N1 HA ( Fig. 1a ). The reemerging swine-origin HA of the 2009 H1N1 pandemic and those that circulated during the 1918–40 period are characterized by AI values > 70% and markedly differ from the strains that circulated during 1940–2008 (varies from 48% to 77% with an average of 55%). Two ‘classical’ swine viruses (data points marked by black arrows), A/New Jersey/76 (H1N1) and A/Wisconsin/4754/1994 (H1N1), isolated between 1940–2008, also have high AI value and are genetically distinct when compared to the main cluster of human influenza viruses circulating in that period. Both viruses are known to have caused human infections following pig-human interspecies transmission. The A/New Jersey/76 influenza virus is reported to have caused respiratory illness in 13 soldiers with 1 death at Fort Dix, New Jersey21. The A/Wisconsin/4754/1994 virus was recovered from a 39 year-old man who came in close contact with experimentally infected pigs22. For the H2 subtype, the HA of the avian H2 strains were compared to the 1957–58 pandemic H2N2 HA (A/Albany/6/58(H2N2)) and the antigenic sites I-A, I-B, I-C, I-D, II-A and II-B23 characterized by hybridoma antibodies generated in BALB/c mice were used to calculate AI. Consistent with the findings of a previous report11, the AI values indicate that the antigenic sites of the 1957–58 pandemic H2N2 HA are conserved in circulating avian H2 influenza viruses ( Fig. 1b ). In fact, the antigenic sites of the majority of avian H2 viruses in circulation are 100% identical to the 1957–58 pandemic H2N2 HA ( Fig. 1b ). The conservation of antigenic sites in swine H2 influenza could not be assessed using this method due to lack of sequence information (H2N2 viruses do not circulate in swine; indeed, infection of swine with H2 viruses is rarely recorded). Similar to H1 subtype, the evolution of human H2 is characterized by steady antigenic drift leaning away from the pandemic strain. Although the majority of the viral strains that circulated during the immediate post-pandemic period 1957–68 have AI values > 70%, viral strains with AI ~ 60% appeared after 1967 ( Fig. 1b ). It is reasonable to expect that the AI values would have decreased further had H2 continued to circulate in human population as a seasonal virus after 1968. The above analyses using H1 and H2 subtypes suggest that viruses carrying pandemic HA-like genes can be distinguished from seasonal viruses using a cutoff value AI ~ 70%.

Trends in antigenic evolution over time.
Antigenic identity of HA from human, avian and swine species relative to pandemics that of note: (a) 1918–19 H1N1, (b) 1957–58 H2N2, (c) 1968–69 H3N2 plotted against time of isolation (x-axis). To generate the plots, 2, 927 human, 166 avian, 950 swine HAs of H1 subtype; 117 human and 163 avian HAs of H2 subtype; 3,632 human, 756 avian and 347 swine HAs of H3 subtype were used. The two black arrows in Fig. 1a correspond to A/New Jersey/76 (H1N1) and A/Wisconsin/4754/1994 (H1N1), both of which caused human infections following pig-human interspecies transmission and have high AI values similar to the 2009 pandemic H1N1 strains. Dotted trendlines are added to graphically display the antigenic drift in avian vs. human H2 (Fig. 1b). The slope of the avian H2 trendline is 0.0845, whereas the slope of the human H2 trendline is −1.866. The dotted horizontal line indicates cutoff AI values (70% (H1); 70% (H2); 70% & 49% (H3)).The data points were jittered slightly on y-axis to avoid large overlaps (AI ~ AI + ε, where −1< ε <1).
In the case of H3, the 5 antigenic sites (A–E)24,25 were used to calculate AI in reference to the prototypic pandemic strain of 1968 (A/Aichi/2/1968 (H3N2)). Unless stated otherwise henceforth an AI value for a given H3 HA sequence refers to its antigenic identity with the 1968 pandemic H3 HA. A total of 1,103 H3 avian and swine sequences were downloaded from the NCBI Influenza Database26 and analyzed. Of these 1,103 sequences, 756 were of avian origin and 347 were of swine origin. The avian sequences comprised nine different subtypes (H3N1-9) and the swine sequences comprised four different subtypes (H3N1, H3N2, H3N3 and H3N8). The avian and swine H3 amino acid sequences were compared against A/Aichi/2/1968 and AI values were computed for all the sequences ( Fig. 1c ). In addition, a total of 3,632 human-adapted H3N2 HA sequences were downloaded from the NCBI database and compared against 1968 pandemic H3 HA to enable a cross-species comparison of the antigenic drift ( Fig. 1c ). The AI values and phylogeny analysis indicate that, in comparison with recent human H3, avian and swine H3 are genetically and antigenically closer to the 1968 pandemic HA. Thus, we confirmed that avian and swine H3 are indeed antigenically intact ( Figs. 1c & S1 ).
In addition to the amino acids that constitute the antigenic sites, the attachment of complex glycans at specific glycosylation sites (Asn-X-Ser/Asn-X-Thr, where X is not a Proline) is also often part of the antigenic surface. An increase or decrease in the number of N-glycosylation sites therefore critically governs the antigenic properties of HA27. The 1968 pandemic H3 HA carries only two glycosylation sites on the globular head region (at 81 & 165), whereas HA from seasonal strains carries an average of six sites (at 63, 122, 126, 133, 144, 165)28. To incorporate glycosylation in the calculation of antigenic identity, the globular head region of the avian and swine HA sequences were examined for the conservation of 1968 pandemic H3-like glycosylation pattern (Methods). Among the 1,103 avian and swine H3 HA sequences, 359 carried additional glycosylation sites or positional shifts and therefore were removed from further consideration. The remaining 744 HA sequences (~ 67%) were found to possess the 1968 pandemic HA-like glycosylation pattern. Out of the 744 HA sequences, strains corresponding to 449 sequences (all avian) were isolated after 2000—many as recently as 2010—and their AI value exceed 70%.
Extrapolating from H1 and H2 pandemic scenarios, the above strains are likely to pose a threat should they acquire the mutations necessary to crossover into human population. Of note, a novel H3N8 avian influenza virus acquired the ability to infect harbor seals in New England recently29. The AI of the seal H3N8 HA is 78%, which is the habitual AI range of avian H3 influenza viruses. Given the high AI value, the history of the spread of avian influenza to humans and the fact that seal H3N8 has already acquired potential to bind sialic acid receptors that are commonly found in the mammalian respiratory tract29, seal H3N8 virus could jump, directly or via reassortment, to humans with pandemic consequences. More recently, the CDC reported the outbreak of a triple reassortant H3N2 swine-origin influenza virus (SOIV) and released a set of sequences at Global Initiative on Sharing All Influenza Data (GISAID) following this event. The HA of a prototype outbreak strain, A/Minnesota/11/2010 (referred as Minn10), shares very high homology (approx. 98%) with the HA of swine A/swine/Minnesota/7931/2007(H3N2) (SwMinn10) and has good binding and transmission properties30. Although the AI value of SwMinn10 (approx. 39%) is comparable to that of a typical seasonal H3 HA, they share very low antigenic identity between them (only 15 out of the 27 [approx. 55%] antigenic positions are conserved). More importantly, the glycosylation pattern appears to be very different between SwMinn10 and seasonal H3 HA. The SwMinn10 HA contains only three glycosylation sites in the globular head region, compared to 6 for the seasonal HA. The swine predecessor was not part of the 581 sequences identified by the analysis. This is due to its low AI value and the extra (third) glycosylation site in the head region. Although Minn10 cannot be regarded as a strain resembling the 1968 pandemic strain, the outbreak caused by this virus supports our theory that avian and swine strains that are divergent enough from the seasonal HA, both antigenically and with respect to their glycosylation pattern, need to be considered as potential threats. Consequently, based on the above observations, we relaxed the criteria used to identify potential pandemic strains and considered those HAs isolated after 2000, having matching glycosylation pattern as pandemic H3 and whose AI was equal to or greater than 49%, the maximum AI value of recent seasonal H3 (2000 or after) ( Fig. 1c ). This yielded 581 sequences (549 avian, 32 swine).
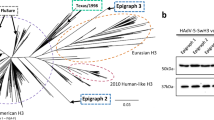
If a virus carrying a HA similar to any one of the 581 sequences acquires the potential to crossover into humans, it would likely have a major impact on both immune recognition and vaccine efficacy. The efficacy of the influenza vaccine in humans is thought to correlate well with the ‘antigenic relatedness’ metric (reciprocal of antigenic distance) obtained from ferret antisera hemagglutinin inhibition (HI) assays31,32 between the vaccine strain and the circulating epidemic strains33,34. We tested the degree of correlation between the AI values and the HI-derived antigenic relatedness to: (1) assess the potential of AI in predicting vaccine-induced cross-reactive antibody responses; and (2) to evaluate the cross-protective capacity of the current vaccine strain, A/Victoria/361/2011 (H3N2), against potential threats. For this exercise, we analyzed three sets of ferret serum HI cross-reactivity data where amino acid sequences of the HA1 polypeptide were present. The first set contained 7 viral strains (21 pairwise comparisons) isolated from 2008 to 201035. The second set contained 9 viral strains (36 pairwise comparisons) isolated from 1970 to 197936. The third set contained 6 viral strains (15 pairwise comparisons) isolated from 1994 to 199937. Antigenic relatedness between two viral strains based on ferret anti-serum was determined using the method described by Lee et al.32. Briefly, the antigenic relatedness between two viral strains is directly proportional to the ratio of the product of the heterologous titers against each other to the product of the homologous titers. In total, 72 pairwise comparisons among 22 viruses were available for analysis. Among the 72 pairwise comparisons, 5 (7%) have an antigenic relatedness > 70% (i.e., similar antigenicity) and 67 (93%) have an antigenic relatedness < 70% (i.e., antigenic variant). Results indicate that the AI values have significant correlation with the HI-based antigenic relatedness metric ( Tables 1 , 2 , 3 ), indicating that AI values could be applied to predict vaccine-induced cross-reactive antibody responses and thus selection of vaccine strains. Particularly, antigenically related viral strains (> 70%) have AI > 80%, hence we employed an 80% cutoff to determine protection, or lack thereof, between a vaccine strain and a challenge viral strain. The current H3N2 vaccine strain A/Victoria/361/2011 has AI values of 92% with the seasonal viral strain A/Brisbane/10/2007, 29% with the A/Aichi/2/68, 56% with a typical H3N2 SOIV and 44% with a representative swine H3 strain from the group of swine viral strains having AI > 49%. These data indicate that the current vaccine strain is unlikely to offer cross-protection against the circulating swine or SOIV viruses whatsoever. Supporting this, IgG polyclonal raised in rabbit with seasonal vaccine H3 strain (A/Brisbane/10/2007(H3N2)) preferentially bind to current seasonal H3 but have weaker affinity to a representative swine H3 ( Fig. 2 ). More significantly, out of the 581 HA sequences, six swine HAs already contain the prototypic mutations (L226, S228) necessary for HA human adaptation38 and are thus capable of entering the human population either directly or via reassortment ( Table 4 , Fig. 3a )38. We recombinantly expressed HA derived from two swine isolates, A/swine/Chonburi/05CB2/2005 (H3N2) and A/swine/Nakhon pathom/NIAH586-2/2005 (H3N2), which have high AI value ( Table 4 ) and characterized their relative binding affinities to representative avian and human receptors on a glycan array platform (Methods, Fig. 4 ). Both swine HAs showed high affinity binding to both human and avian receptors. The high affinity human receptor-binding of these swine HAs appears to be in the same range as that of other seasonal H3 HAs characterized previously39,40 and are thus capable of entering the human population either directly or via reassortment. The antigenic relationship of these HAs (AI value and glycosylation pattern) to the pandemic 1968 H3N2 HA strongly suggests that the six isolates belong to swine virus lineage and not examples of transient reverse zoonoses. Phylogenetic analysis of the 32 swine isolates revealed that majority of them fall under European and Asian swine lineages.

Binding of anti-A/Brisbane/10/2007(H3N2) pAb to H3 strains measured by ELISA.
Tested were 2 swine H3N2 HAs (A/swine/Chonburi/05CB2/2005, A/swine/Nakhon pathom/NIAH586-2/2005), pandemic H3N2 HA (A/Aichi/2/1968), 1 seasonal H3N2 HA (A/Wisconsin/67/2005). The seasonal vaccine H3N2 HA (A/Brisbane/10/2007) and a representative H7N7 HA (A/Netherlands/219/2003) were used as positive and negative controls, respectively.

Genetic, antigenic and glycosylation-pattern relatedness of 1968 pandemic H3N2 HA to seasonal, swine and avian H3 HA.
(a) Sequence alignment of the expanded globular head region (residues 50–328) of the HAs listed in Table 4 . Antigenic sites A, B, C, D, E of H3 HA are highlighted in green, magenta, cyan, grey and yellow, respectively. In each sequence, the Asn residue associated with the N-linked glycosylation sites (Asn-X-Ser/Asn-X/Thr) is marked in red. (b) Surface rendered three-dimensional structural models of trimeric HA1 globular head of representative pandemic (middle), seasonal (right) and swine (left) HAs. The view of trimer is along axis perpendicular to 3-fold symmetry axis to give a complete picture of the antigenic and glycosylation sites. The homology models of HA1 chain were generated using SWISS-MODEL automated modeling server (http://swissmodel.expasy.org/) and the trimannosyl N-linked glycosylation at the sites were added in silico using GlyProt server (http://www.glycosciences.de/modeling/glyprot/php/main.php). The trimeric HA1 was generated by superimposing three copies of homology-modeled glycosylated monomer with corresponding monomers in the trimer crystal structure (PDB ID: 1HGE). The antigenic sites A–E are marked on the structure. With 1968 pandemic HA as reference (antigenic sites shown in red), the structural similarity of the antigenic sites in seasonal and swine HAs to the reference HA is shown in different shades of red (duller shade representing low similarity to brighter shade representing high similarity).

Glycan microarray analysis of representative H3 HAs.
Dose dependent binding of A/swine/Nakhon pathom/NIAH586-2/2005 (a) and A/swine/Chonburi/05CB2/2005 (b) HAs to representative avian and human glycan receptors on the glycan array platform is shown. Both these HAs show high affinity binding to both human receptors (6′SLN-LN) and avian receptors (3′SLN-LN and 3′SLN-LN-LN).
The analyses presented here portend a vaccine strategy to prevent a future H3 pandemic. Among the WHO recommended vaccine strains of influenza A/H3N2 virus, A/Hong Kong/1/1968(H3N2) will be effective (AI > 80%) against 505 of 581 strains (~ 87%) identified by this study and thus could be used for the development of pandemic influenza vaccine. Surprisingly, H3N2 vaccine strains that were subsequently used are not capable of being as effective. These data suggest that a cocktail of A/Hong Kong/1/1968(H3N2) and an avian and swine strain each that represent the circulating influenza in birds and pigs can form the components of the pandemic influenza vaccine.
To understand the results from AI calculations in the context of the spatial relationship between glycosylation site and antigenic sites of H3 HA we constructed structural homology models of HA1 globular head of ACHI68, BRBN07 and CHIB05 HAs (see Table 4 for strain information). These structural models of HA comprised the basic trimannosyl core of N-linked glycan attached to the glycosylation sites ( Fig. 3b ). From the structural comparison it is clear that antigenic shape of HA which includes antigenic sites A-E and the glycosylation pattern of HA1 from the swine strain (CHIB05) closely resembles that of the 1968 pandemic HA. Conversely, the antigenic shape of a more recent seasonal strain (BRBN07) is remarkably different from that of the pandemic strain.
Discussion
The H3 HA of some of the recent avian strains share approximately 86% overall sequence identity with the HA of the avian progenitor of the 1968 pandemic virus (A/duck/Ukraine/1/1963), reflecting antigenic intactness within birds. Many sequences from swine, some collected as recently as 2001, were also found to have high homology with the A/duck/Ukraine/1/1963 HA, indicating avian to swine transfer. For reasons that remain unclear, the more recent swine H3 HAs (2006 and later) have diverged significantly from the 1968 pandemic H3N2 HA ( Fig. 1c ) while in contrast the majority of swine H1 HAs remained antigenically stable from 1918 to the 1990s. Unlike the 1918 H1N1 virus which crossed to swine soon after and remained in swine, the human H3N2 viruses have repeatedly crossed from humans to swine for some time – quite possibly, this could be the reason why swine H3 viruses appear to manifest the antigenic drift that human strains underwent during this period. In fact, the AI values of human H3 in the last decade are comparable to the AI values of some swine H3 HAs of the same period; interestingly, the recent human and swine HAs show differential binding to polyclonal antibodies generated against seasonal vaccine strain ( Fig. 2 ). This apparent discrepancy may be explained in part by the presence of certain key antigenic "hotspot" locations, where amino acid substitutions can lead to disproportionately large changes in antigenicity. Our observation is supported by other studies on H3 antigenic evolution14,41. The frequent interspecies transmission of H3 viruses might also explain why this subtype is associated with the highest rates of mortality42.
The importance of glycosylation in antigenic site masking leading to a new pandemic cycle and viral evolution became apparent after the 2009 pandemic. It was observed that the seasonal H1N1 HA carries antigenic site-masking glycosylation sites not present in the 2009 pandemic H1N1 HA (and 1918 H1N1 HA) and the exposure of the unprotected antigenic surface is believed to be the reason underpinning the severity of the 2009 H1N1 pandemic. Akin to H1 subtype, the additional glycosylation sites on the recent seasonal H3 appear to have a role in antigenic site-masking. For instance, the glycosylation at position 63 masks antigenic site E and glycosylation at sites 122, 133 and 144 protect antigenic A. The shielding nature of these glycosylation sites is evident from the gradual decline in the mutation rate of the masked antigenic sites following their appearance ( Fig. S2 ), portending a 2009 H1N1-like H3N2 pandemic. If a virus carrying a HA similar to the ones identified by this analysis makes its way into humans, it would need to evolve rapidly in response to selective pressures from vaccination and herd immunity. The ability of H3 subtype to add glycosylation sites will be a key factor enabling the virus to achieve sustained circulation in the next cycle. In contrast, a previous study43 using nucleotide sequence analysis concluded that H2 has an intrinsically lower capacity to add glycosylation sites. Taking these factors together, we assert that it is less likely for an avian or swine H2 virus (antigenically similar to 1957–58 pandemic H2N2) to gain a foothold for sustained circulation in humans when compared to H3 viruses.
The rapid antigenic drift that human H3N2 HA underwent during the early adaptation period of the virus (1968–76) appears to have slowed down after 1977 ( Fig. 1c ). Interestingly, this time period also coincides with the reemergence of H1N1 in the human population. The (re-) emerging H1N1 subtype could have imposed strong selective pressures on the H3N2 to stop circulating in humans after 1977. The evolution of human H3N2 HA after 1977 is characterized by glycosylation accrual, low-level site-specific antigenic changes and variations at other non-immunodominant sites ( Fig. 1c ). Additionally, a recent study found that the affinity of human H3 viruses for human receptors has reduced drastically since 200144. These observations suggest that currently circulating viruses are not as dominant as the earlier viruses. Based on this trend, one can argue that human H3N2 HA presently is “antigenically drained”, which poses a substantially high barrier to evolution via antigenic drift. However, the presence of antigenically intact H3 in avian and swine suggests that, as with 2009 H1N1 pandemic, reassortment can result in ‘resetting and shifting’ the antigenicity back to that of the 1968 pandemic and hence facilitate sustained evolution of this subtype in humans.
Influenza A viruses of other subtypes (H5, H7, H9) that have caused sporadic infections in humans over the past decade also pose equal risk of a pandemic, especially since they represent completely novel HA subtypes. Although antigenic phenotypes could be predicted from HA sequences, the genetic signatures in influenza viruses that lead to a sustained human-to-human transmission cannot be accurately predicted. Although an antigenically novel HA is necessary, it is not the only determining factor for a pandemic. While gain of host receptor specificity is a key determinant, changes in influenza proteins other than HA such as the polymerase (PB2) are typically involved, making predictions of the timing of future pandemics more complex. Nevertheless, our study facilitates setting the stage for future work aimed at designing vaccination studies with animal models using a cocktail of H3 antigens from strains of current avian and swine origin along with specific past strains. Such studies would augment the preparedness in the event of potential re-emergence of H3N2 pandemic45.
Methods
Calculation of AI values for H1, H2 and H3 subtypes
AI values were calculated using the characterized antigenic sites of H1, H2 and H3 HA. For H1, 128, 129, 158, 160, 162, 163, 165, 166, 167 (Sa); 156, 159, 192, 193, 196, 198 (Sb); 140, 143, 145, 169, 173, 182, 207, 224, 225, 240, 273 (Ca); 78, 79, 81, 82, 83, 122 (Cb) were used. For H2, 162, 248 (I-A); 137, 187 (I-B); 131, 222, 218 (I-C), 80, 200 (I-D); 40 (II-A), 273 (II-B) were used. For H3, 122, 133, 137, 143, 144, 145, 146 (A); 155, 186, 188, 189, 193 (B); 53, 54, 275, 278 (C); 201, 205, 207, 208, 217, 220 (D); 62,78,81,83 (E) were used. Positions are numbered according to H3 molecule. The antigenic identity (AI) of an avian or a swine HA is defined by the percentage fraction of amino acids in the dominant antigenic sites that are conserved in the corresponding pandemic HA for each of the H1 (A/South Carolina/1/18), H2 (A/Albany/6/58(H2N2)) and H3 (A/Aichi/2/1968 (H3N2)) subtypes.
In silico identification of glycosylation sites
Glycosylation sites are defined by the motif N-X-T/S, where X is any amino acid except Proline. A position in a HA amino acid sequence is considered to be glycosylated if it contains the N-X-T/S motif and is predicted by GlyProt (http://www.glycosciences.de/modeling/glyprot/php/main.php) – an online tool for in silico glycosylation of proteins.
Cloning, baculovirus synthesis, recombinant expression and purification of representative H3 Has
Soluble versions (lacking membrane proximal C-terminus region) of HA from representative H3N2 swine isolates A/swine/Chonburi/05CB2/2005 and A/swine/Nakhon pathom/NIAH586-2/2005 were recombinantly expressed (with C-terminal His-tag) as described previously40. These representative H3 HAs had high AI values and the prototypic Leu226 and Ser228 residues characteristic of human-adapted H3 HAs. Briefly, recombinant baculoviruses with the HA gene were used to infect (MOI = 1) suspension cultures of Sf9 cells (Invitrogen, Carlsbad, CA) cultured in BD Baculogold Max-XP SFM (BD Biosciences, San Jose, CA). The infection was monitored and the conditioned media was harvested 3–4 days post-infection. The soluble HA from the harvested conditioned media was purified using Nickel affinity chromatography (HisTrap HP columns, GE Healthcare, Piscataway, NJ). Eluting fractions containing HA were pooled, concentrated and buffer exchanged into 1X PBS pH 8.0 (Gibco) using 100K MWCO spin columns (Millipore, Billerica, MA). The purified protein was quantified using BCA method (Pierce).
Glycan array analysis
To investigate the multivalent HA-glycan interactions a streptavidin plate array comprising of representative biotinylated α2→3 and α2→6 sialylated glycans was used as described previously40. 3′SLN, 3′SLN-LN, 3′SLN-LN-LN are representative avian receptors. 6′SLN and 6′SLN-LN are representative human receptors. The biotinylated glycans were obtained from the Consortium of Functional Glycomics through their resource request program. Streptavidin-coated High Binding Capacity 384-well plates (Pierce) were loaded to the full capacity of each well by incubating the well with 50 μl of 2.4 μM of biotinylated glycans overnight at 4°C. Excess glycans were removed through extensive washing with PBS. The trimeric HA unit comprises of three HA monomers (and hence three RBS, one for each monomer). The spatial arrangement of the biotinylated glycans in the wells of the streptavidin plate array favors binding to only one of the three HA monomers in the trimeric HA unit. Therefore in order to specifically enhance the multivalency in the HA-glycan interactions, the recombinant HA proteins were pre-complexed with the primary and secondary antibodies in the molar ratio of 421 (HA: primary: secondary). The identical arrangement of 4 trimeric HA units in the pre-complex for all the HAs permit comparison between their glycan binding affinities. A stock solution containing appropriate amounts of Histidine tagged HA protein, primary antibody (Mouse anti 6X His tag IgG) and secondary antibody (HRP conjugated goat anti Mouse IgG (Santacruz Biotechnology) in the ratio 4:2:1 and incubated on ice for 20 min. Appropriate amounts of pre-complexed stock HA were diluted to 250 μl with 1% BSA in PBS. 50 μl of this pre-complexed HA was added to each of the glycan-coated wells and incubated at room temperature for 2 hours followed by the above wash steps. The binding signal was determined based on HRP activity using Amplex Red Peroxidase Assay (Invitrogen, CA) according to the manufacturer's instructions. The experiments were done in triplicate. Minimal binding signals were observed in the negative controls including binding of pre-complexed unit to wells without glycans and binding of the antibodies alone to the wells with glycans.
References
Cunha, B. A. Influenza: historical aspects of epidemics and pandemics. Infectious disease clinics of North America 18, 141–155 (2004).
Russell, C. J. & Webster, R. G. The genesis of a pandemic influenza virus. Cell 123, 368–371 (2005).
Taubenberger, J. K. et al. Characterization of the 1918 influenza virus polymerase genes. Nature 437, 889–893 (2005).
Scholtissek, C., Rohde, W., Von Hoyningen, V. & Rott, R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 87, 13–20 (1978).
Kawaoka, Y., Krauss, S. & Webster, R. G. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J Virol 63, 4603–4608 (1989).
Webster, R. G., Bean, W. J., Gorman, O. T., Chambers, T. M. & Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol Rev 56, 152–179 (1992).
Settembre, E. C., Dormitzer, P. R. & Rappuoli, R. H1N1: can a pandemic cycle be broken? Science translational medicine 2, 24ps14 (2010).
Taubenberger, J. K., Reid, A. H. & Fanning, T. G. The 1918 influenza virus: A killer comes into view. Virology 274, 241–245 (2000).
Taubenberger, J. K., Reid, A. H., Janczewski, T. A. & Fanning, T. G. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 356, 1829–1839 (2001).
Gorman, O. T., Bean, W. J. & Webster, R. G. Evolutionary processes in influenza viruses: divergence, rapid evolution and stasis. Curr Top Microbiol Immunol 176, 75–97 (1992).
Nabel, G. J., Wei, C. J. & Ledgerwood, J. E. Vaccinate for the next H2N2 pandemic now. Nature 471, 157–158 (2011).
Smith, G. J. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009).
Cherry, J. L., Lipman, D. J., Nikolskaya, A. & Wolf, Y. I. Evolutionary dynamics of N-glycosylation sites of influenza virus hemagglutinin. PLoS currents 1, RRN1001 (2009).
Smith, D. J. et al. Mapping the antigenic and genetic evolution of influenza virus. Science 305, 371–376 (2004).
Russell, C. A. et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 320, 340–346 (2008).
Jansen, A. G., Sanders, E. A., Hoes, A. W., van Loon, A. M. & Hak, E. Influenza- and respiratory syncytial virus-associated mortality and hospitalisations. Eur Respir J 30, 1158–1166 (2007).
Iwane, M. K. et al. Population-based surveillance for hospitalizations associated with respiratory syncytial virus, influenza virus and parainfluenza viruses among young children. Pediatrics 113, 1758–1764 (2004).
Simonsen, L. et al. Impact of influenza vaccination on seasonal mortality in the US elderly population. Arch Intern Med 165, 265–272 (2005).
Caton, A. J., Brownlee, G. G., Yewdell, J. W. & Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31, 417–427 (1982).
Gerhard, W., Yewdell, J., Frankel, M. E. & Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 290, 713–717 (1981).
Gaydos, J. C., Top, F. H., Jr, Hodder, R. A. & Russell, P. K. Swine influenza a outbreak, Fort Dix, New Jersey, 1976. Emerg Infect Dis 12, 23–28 (2006).
Wentworth, D. E., McGregor, M. W., Macklin, M. D., Neumann, V. & Hinshaw, V. S. Transmission of swine influenza virus to humans after exposure to experimentally infected pigs. J Infect Dis 175, 7–15 (1997).
Tsuchiya, E. et al. Antigenic structure of the haemagglutinin of human influenza A/H2N2 virus. J Gen Virol 82, 2475–2484 (2001).
Wiley, D. C., Wilson, I. A. & Skehel, J. J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Wilson, I. A., Skehel, J. J. & Wiley, D. C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289, 366–373 (1981).
Bao, Y. et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol 82, 596–601 (2008).
Xu, R. et al. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328, 357–360 (2010).
Zhang, M. et al. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV and HCV envelopes and influenza hemagglutinin. Glycobiology 14, 1229–1246 (2004).
Anthony, S. J. et al. Emergence of fatal avian influenza in New England harbor seals. mBio 3, e00166–00112 (2012).
Pearce, M. B. et al. Pathogenesis and transmission of swine origin A(H3N2)v influenza viruses in ferrets. Proc Natl Acad Sci U S A 109, 3944–3949 (2012).
Smith, D. J., Forrest, S., Ackley, D. H. & Perelson, A. S. Variable efficacy of repeated annual influenza vaccination. Proc Natl Acad Sci U S A 96, 14001–14006 (1999).
Lee, M. S. & Chen, J. S. Predicting antigenic variants of influenza A/H3N2 viruses. Emerg Infect Dis 10, 1385–1390 (2004).
Klimov, A., Simonsen, L., Fukuda, K. & Cox, N. Surveillance and impact of influenza in the United States. Vaccine 17 Suppl 1, S42–46 (1999).
WHO Recommended composition of influenza virus vaccines for use in the 2013 southern hemisphere influenza season. Wkly Epidemiol Rec 87, 389–400 (2012).
WHO Antigenic and genetic characteristics of zoonotic influenza viruses and development of candidate vaccine viruses for pandemic preparedness. Wkly Epidemiol Rec 86, 469–480 (2011).
Both, G. W., Sleigh, M. J., Cox, N. J. & Kendal, A. P. Antigenic drift in influenza virus H3 hemagglutinin from 1968 to 1980: multiple evolutionary pathways and sequential amino acid changes at key antigenic sites. J Virol 48, 52–60 (1983).
Coiras, M. T. et al. Rapid molecular analysis of the haemagglutinin gene of human influenza A H3N2 viruses isolated in spain from 1996 to 2000. Arch Virol 146, 2133–2147 (2001).
Connor, R. J., Kawaoka, Y., Webster, R. G. & Paulson, J. C. Receptor specificity in human, avian and equine H2 and H3 influenza virus isolates. Virology 205, 17–23 (1994).
Chandrasekaran, A. et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol 26, 107–113 (2008).
Srinivasan, A. et al. Quantitative biochemical rationale for differences in transmissibility of 1918 pandemic influenza A viruses. Proc Natl Acad Sci U S A 105, 2800–2805 (2008).
de Jong, J. C. et al. Antigenic and genetic evolution of swine influenza A (H3N2) viruses in Europe. J Virol 81, 4315–4322 (2007).
Thompson, W. W. et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 (2003).
Igarashi, M., Ito, K., Kida, H. & Takada, A. Genetically destined potentials for N-linked glycosylation of influenza virus hemagglutinin. Virology 376, 323–329 (2008).
Lin, Y. P. et al. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc Natl Acad Sci U S A 109, 21474–21479 (2012).
Settembre, E. C., Dormitzer, P. R. & Rappuoli, R. H1N1: can a pandemic cycle be broken? Sci Transl Med 2, 24ps14 (2010).
Acknowledgements
This work was funded in part by National Institutes of Health (R37 GM057073-13) and the National Research Foundation supported Interdisciplinary Research group in Infectious Diseases of SMART (Singapore MIT alliance for Research and Technology).
Author information
Authors and Affiliations
Contributions
K.T., R.R., V.S. and R.S. conceptualized and designed the study. K.T. implemented the AI method and sequence analysis. R.R. performed the structural modeling. K.V. and N.W.S. designed and performed the experiments on glycan array analysis and Ig cross reactivity. K.T., R.R., V.S. and R.S. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tharakaraman, K., Raman, R., Stebbins, N. et al. Antigenically intact hemagglutinin in circulating avian and swine influenza viruses and potential for H3N2 pandemic. Sci Rep 3, 1822 (2013). https://doi.org/10.1038/srep01822
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01822
This article is cited by
-
A multifunctional human monoclonal neutralizing antibody that targets a unique conserved epitope on influenza HA
Nature Communications (2018)
-
H5N1 influenza vaccine induces a less robust neutralizing antibody response than seasonal trivalent and H7N9 influenza vaccines
npj Vaccines (2017)
-
Quantifying influenza virus diversity and transmission in humans
Nature Genetics (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
