
Abstract
A series of edge-selectively halogenated (X = Cl, Br, I) graphene nanoplatelets (XGnPs = ClGnP, BrGnP, IGnP) were prepared simply by ball-milling graphite in the presence of Cl2, Br2 and I2, respectively. High BET surface areas of 471, 579 and 662 m2/g were observed for ClGnP, BrGnP and IGnP, respectively, indicating a significant extent of delamination during the ball-milling and subsequent workup processes. The newly-developed XGnPs can be well dispersed in various solvents and hence are solution processable. Furthermore, XGnPs showed remarkable electrocatalytic activities toward oxygen reduction reaction (ORR) with a high selectivity, good tolerance to methanol crossover/CO poisoning effects and excellent long-term cycle stability. First-principle density-functional calculations revealed that halogenated graphene edges could provide decent adsorption sites for oxygen molecules, in a good agreement with the experimental observations.
Similar content being viewed by others

Introduction
One of the major hurdles for commercialization of the fuel cell technology is the sluggish oxygen reduction reaction (ORR) at cathode1,2,3. So far, high cost and scarce precious platinum (Pt) and its alloys have been considered to be the most reliable cathodic ORR electrocatalysts in fuel cells4,5,6,7,8. In addition to the high cost, however, Pt and its alloys are also suffered from methanol crossover/carbon monoxide (CO) poisoning effects and poor operation stability. Therefore, it is essential to search for non-precious metal9,10,11 or metal-free12,13,14,15,16,17 electrocatalysts with a high catalytic activity and long-term operation stability to reduce or replace Pt-based ORR electrocatalysts in fuel cells. Although extensive efforts have been devoted to the development of non-precious metal-based electrocatalysts, their practical application is still out of sight due largely to their limited electrocatalytic activity, poor cycle stability and sometimes environmental hazard.
Recently, carbon-based materials doped with heteroatoms, such as boron (B)14,18, halogen (Cl, Br, I)19,20, nitrogen (N)12,15,21,22,23,24,25, phosphorus (P)26, sulfur (S)27 and their mixtures28,29,30, have attracted tremendous attentions as metal-free ORR eletrocatalysts. The difference in electronegativity (χ) between the heteroatom dopants (B = 2.04, I = 2.66, N = 3.04, P = 2.19 and S = 2.58) and carbon atom (2.55)31 in covalently doped graphitic carbon frameworks can polarize adjacent carbon atoms. Indeed, quantum mechanics calculations revealed that the electron accepting/donating ability of the heteroatom dopants created net positive/negative charges on adjacent carbon atoms in graphitic lattice to facilitate the oxygen reduction process12. Thus, both the vertically-aligned nitrogen-doped carbon nanotubes (VA-NCNTs)12 and nitrogen-doped graphene (N-graphene)25 catalyzed an efficient four-electron ORR process with a higher electrocatalytic activity and better operation stability than the commercially available Pt/C-based electrocatalyst (Pt: 20 wt%, Vulcan XC-72R). Furthermore, the excellent stability over the methanol crossover/CO poisoning effects is additional advantage of these carbon-based metal-free catalysts. Although the basic catalytic mechanism has been established, the full potential of these carbon-based, metal-free catalysts is hard to achieve without the synthetic capability for large-scale production of the heteroatom-doped, carbon-based materials at low cost. However, commonly affordable chemical vapor deposition (CVD)12,14 for the preparation of carbon nanotubes and graphene sheets and/or Hummers' methods for graphene oxide production are too expensive and involve environmentally hazardous reagents and thus inappropriate for large-scale production32.
In this study, we have, for the first time, synthesized a series of edge-selectively halogenated (Cl, Br and I) graphene nanoplatelets (ClGnP, BrGnP and IGnP; collectively designated as XGnPs) by simply ball-milling graphite flake33 in the presence of chlorine (Cl2), bromine (Br2) or iodine (I2), respectively. Since fluorine (F2) gas is too reactive and toxic to be handled in normal laboratories (serious cautions!)34,35, edge-fluorinated GnP (FGnP) could not be prepared for this study. Although the electronegativities of halogen atoms are in the order of Cl (3.16) > Br (2.96) > I (2.66), the experimentally observed electrocatalytic activities for ORR are in the order of IGnP > BrGnP > ClGnP. Compared to the pristine graphite and commercially available Pt/C electrocatalyst, IGnP exhibited much better electrocatalytic performances for ORR with an excellent long-term durability and tolerance to methanol crossover/CO poisoning effects. The density-functional theory (DFT) calculations revealed that certain particular types of halogenated edges showed desirable binding affinity with oxygen (O2) molecules. In consistency with the experimental observations, the charge-transfer induced weakening of the O-O bond strength was most prominent for the case of IGnP.
Results
The ball-milling-driven mechanochemical reaction between active carbon species and halogens is schematically shown in Figure 1a. Briefly, the high speed rotation (500 rpm) of the stainless steel balls during ball milling generated sufficient kinetic energy to cause bond cleavages for the graphitic C-C framework (Figure 1b). As a result, active carbon species (mostly carboradicals, carbocations and carbanions)36 formed at the broken edges of graphite, which were sufficiently reactive to pick up halogens (e.g., Cl2, Br2, I2) in the sealed ball-mill capsule. The detailed mechanism is proposed in Figure S1 in the Electronic Supporting Information (ESI). The high reactivity of the active carbon species was indicated by violent sparkling observed when capsule lid was opened (Figures 1c, 1d and supporting video clip in ESI), presumably due to the termination reaction for the remnants of the active carbon species with air moisture.

(a) A schematic representation for mechanochemically driven edge-halogenation reaction between the in-situ generated active carbon species (gold balls) and reactant halogens (twin green balls). Active carbon species were generated by homolytic bond cleavages of graphitic C-C bonds and reacted with halogen molecules to produce edge-halogenated graphene nanoplatelets (XGnPs) in a sealed ball-mill capsule and the remnant active carbon species are terminated upon subsequent exposure to air moisture. Red and gray balls stand for oxygen and hydrogen, respectively; (b) ball-mill capsule containing the pristine graphite and stainless steel balls (diameter 5 mm); (c) violent sparkling (red spots) occurred when the reaction mixture was exposed to ambient air moisture and excess purple I2 was sublimated in the air (arrow); (d) continued sparkling from residual IGnP at the bottom of a ball-mill capsule even after most of the IGnPs and stainless balls were taken out. The images were captured from supporting video clip in ESI.
Scanning electron microscopy (SEM) images show an obvious size reduction from a large grain size of :150 μm for the pristine graphite (Figure 2a) to a much smaller grain size of <1 μm for XGnPs (Figures 2b–2d) by ball-milling. The size reduction implies C-C bond breakings in the graphitic structures and thus in-situ generation of active carbon species to react with halogens. The presence of Cl, Br and I in the resultant XGnPs are clearly evident by energy dispersive X-ray (EDX) spectroscopic (Figure 2e) measurements with element mapping (Figure S2 and Table S2) and elemental analyses (EA) (Table S1).

SEM images.
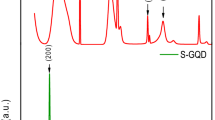
(a) the pristine graphite with average grain size :150 μm. The average grain sizes of XGnPs reduced to less than 1 μm: (b) ClGnP; (c) BrGnP; (d) IGnP. Scale bars are 1 μm. (e) EDX spectra. (f) XRD diffraction patterns. Inset is magnified XRD diffraction patterns from the pink rectangle. The relative [002] peak intensities of XGnPs are less than 0.4% of that of the pristine graphite, indicating that the great extent of graphite has been deliminated into edge-halogenated graphene nanoplatelets (XGnPs). (g) A schematic representation for the edge expansions of XGnPs caused by the edge-halogens.
XRD diffraction patterns are shown in Figure 2f. As can be seen, the pristine graphite exhibited a typical strong [002] peak at 26.5°, corresponding to an interlayer d-spacing of 0.34 nm37. In contrast, all XGnPs displayed less than 0.4% of the [002] peak intensity characteristic of the pristine graphite, suggesting a high degree of exfoliation occurred during the ball-milling and subsequent work-up processes. Unlike graphite oxide (GO) with a large parallel shift of the [002] peak to as low as 10.5° (d-spacing of 0.83 nm)38, XGnPs showed a dramatic decrease in the peak intensity while maintaining the peak location close to 26.5°, indicating that the great extent of graphitic layers were delaminated into XGnPs without much lattice expansion or basal plane damage during the ball milling. As atomic size decreased in the order of I > Br > Cl (Figure 2g), the peaks (d-spacing) appear at 25.3 (0.35 nm), 24.8 (0.36 nm) and 24.1° (0.37 nm) for ClGnP, BrGnP and IGnP, respectively (Figure 2f, inset). The average number of layers in the delaminated graphite by ball-milling could be estimated on the basis of surface area of XGnPs. Given that the maximum BET surface of single layer graphene is 2630 m2/g 39, the average layer numbers of the XGnP samples could be calculated by simply dividing the maximum surface area by experimentally determined BET surface areas of the XGnPs (Table S3). Because the edge contribution of the graphene nanoplatelets (GnPs) is negligible in respect to the basal area, the estimated average numbers of the delaminated graphitic layers are 5.6 (2630/471), 4.5 (2630/579) and 4.0 (2630/662) for ClGnP, BrGnP and IGnP, respectively. Hence, the ball-milling process involves not only mechanochemically cracking graphitic C-C bonds and edge-selectively functionalizing graphitic layers, but also delaminating graphite into GnPs. The XGnPs could be further exfoliated into even fewer-layered GnPs via edge-opening upon dispersion in various polar solvents (Figures S3a–S3c). Zeta-potential data for the XGnPs at different concentrations in DMF (Figures S3d and S3e) are in the range of −31.6–−38.7 mV, indicating the formation of stable dispersions as an absolute Zeta-potential value larger than 30 mV can ensure a stable dispersion via either positive or negative charge repulsion (Table S4)40. Thus, the Zeta-potential measurements, together with microscopic (see Figures 2a–2d) and spectroscopic studies (see Figures 2e and 2f), indicate that the driving force for the good dispersion stability must be originated from the size reduction (entropic contribution) and the steric repulsion between the halogen groups at the edges (enthalpic contribution, Figure 2g)41.
Typical TEM images (Figures S4–S6) with selected area electron diffraction (SAED) patterns (insets, Figures S4a, S5a and S6a) of XGnPs show flake-like morphology with ordered structures at the basal planes and some structural distortions at the edges (insets, Figures S4d, S5d and S6d). Once again, these results confirm the edge-selective halogenations of GnPs by ball-milling with minimal alteration of their basal planes. Element mappings of multilayer XGnPs through the bulk thickness further confirm the presence of halogen heteroatoms in XGnPs (Figures S4e–S4g, S5e–S5g and S6e–S6g).
The degree of halogenations could be quantitatively measured by thermogravimetric analyses (TGA) in nitrogen. The weight losses of ClGnP, BrGnP and IGnP at 800°C were found to be 14.1, 28.6 and 24.6 wt%, respectively, while the pristine graphite displayed a negligible weight loss (0.3 wt%) under the same condition (Figure S7a and Table S2). The major part of the lost weight came from thermal decomposition of edge-halogen groups in XGnPs42 in consistency with the EA data (Table S1).
Raman spectra given in Figure S7b show that the pristine graphite with the relatively large grain size has no detectable D band at around 1350 cm−1 and hence the ratio of the D-band to G-band intensities (ID/IG) is nearly zero. However, all XGnPs showed strong D bands around 1350 cm−1 with the ID/IG ratios in the range of 1.05–1.31, indicating significant size reduction and edge distortion as also indicated by Figures 2b–2d. It is of interest to note that the relative intensity of the D band increased with decreasing atomic size of halogen in the order of I > Br > Cl, accompanied by peak narrowing. The observed peak narrowing can be attributed to the dopant size effect with the bigger edge-functional groups for a more significant graphitic lattice distortion and hence peak broadening, as schematically shown in Figure 2g. The increase in the ID/IG ratios with decreasing atomic size of halogen indicates that the smaller halogen atom is the more number of the atoms has attached onto the edge of XGnPs (vide infra).
To characterize chemical compositions of the as-prepared XGnPs, we performed X-ray photoelectron spectroscopic (XPS) measurements (Figures S7c–S7f). Along with the O1s and C1s peaks, the XPS survey spectra given in Figure S7c clearly show the Cl2p (Figure S7d), Br3d (Figure S7e)43 and I3d (Figure S7f)44 peaks for ClGnP, BrGnP and IGnP, respectively. It was found that the contents of Cl, Br and I to be 5.89, 2.78 and 0.95 at% for ClGnP, BrGnP and IGnP, respectively (see Table S2). In view of its surface sensitive nature, the XPS result may be not always in a good agreement with the EA (see Table S1) and TGA (see Table S2) data. The high-resolution Cl2p (Figure S7d), Br3d (Figure S7e)43 and I3d (Figure S7f)44 spectra show two different types of bonding for all the halogen dopants, leading to the unusual ORR activity observed for XGnPs (vide infra).
Discussion
Having characterized the morphological and the chemical structures for XGnPs, we further investigated their electrocatalytic activities in N2- and O2-saturated 0.1 M aq. KOH solutions using the pristine graphite and commercial Pt/C electrocatalysts with the same mass loadings as reference. Cyclic voltammograms (CV) in Figures 3a–3d show the obvious oxygen reduction peaks for all the four carbon-based electrodes in the O2-saturated 0.1 M aq. KOH solution, while they displayed featureless CVs in the corresponding N2-satureted medium. Figure 3a shows a single cathodic reduction peak at −0.37 V with a current density of −0.28 mA cm−2 for the pristine graphite electrode in the O2-saturated 0.1 M aq. KOH solution. The corresponding cathodic reduction peaks for the ClGnP, BrGnP and IGnP were positively shifted to −0.24, −0.22 and −0.22 V, respectively. The corresponding peak currents for oxygen reduction were determined to be −0.39, −0.60 and −0.78 mA cm−2 for the ClGnP, BrGnP and IGnP, respectively. These values are over 1.4, 2.1 and 2.8 times that of the pristine graphite (−0.28 mA cm−2). Thus, the reduction currents of XGnPs gradually increased while their onset potentials positively shifted along the order of ClGnP < BrGnP < IGnP. As shown in Figures S8–S10 and Table S5, the capacitances of XGnPs gradually increased in the order of the pristine graphite < ClGnP < BrGnP < IGnP. Among all the electrodes tested in this study, the IGnP electrode has the highest capacitances of 127.6 and 139.5 F g−1 in both N2- and O2-saturated electrolytes with a high cycle stability. Clearly, therefore, the edge-halogenations, particularly Br and I, play an important role to significantly improve the ORR activity of graphite, while edge-hydrogenated GnPs (HGnPs), prepared by ball-milling under the same conditions in the absence of halogen, showed marginally improved ORR activity45.

Cyclic voltammograms (CV) of samples on glassy carbon (GC) electrodes in N2- and O2-saturated 0.1 M aq. KOH solution with a scan rate of 10 mV s−1.
(a) the pristine graphite; (b) ClGnP; (c) BrGnP; (d) IGnP; (e) Pt/C. Pink arrows indicate the contributions of hydrogen evolution at around −0.7 V and out of limiting potential (−0.8 V). (f) Linear sweep voltammograms (LSV) at a rotation rate of 1600 rpm and a scan rate of 10 mV s−1, showing a gradual increase in current and a positive shift in the onset potential along the order of the pristine graphite < ClGnP < BrGnP < IGnP < Pt/C (pink arrow).
To further study the ORR performance, we carried out the linear sweep voltammetry (LSV) measurements on a rotating disk electrode (RDE) for the XGnP electrodes, along with the pristine graphite and commercial Pt/C electrodes, in O2-saturated 0.1 M KOH at a scan rate of 0.01 V s−1 and a rotation rate of 1600 rpm. As shown in Figure 3f, the onset potential for oxygen reduction at the pristine graphite electrode is about −0.33 V, which positively shifted to the range of −0.16–−0.14 V upon the edge-functionalization with halogen atoms - though still smaller than that of the Pt/C (−0.06 V). The limiting diffusion currents at −0.8 V for the pristine graphite, ClGnP, BrGnP, IGnP and Pt/C electrodes are −0.09, −0.18, −0.28, −0.40 and −0.30 mA, respectively. These current values for the XGnPs are about 2, 3.1 and 4.4 times higher than that of the pristine graphite and 60%, 93% and 133% that of the Pt/C. These results are consistent with the CV measurements (see Figures 3a–3e and Table S5), confirming, once again, the significant contributions to the ORR electrocatalytic activity of GnPs by the edge-halogenation.
To gain further insight into the kinetics of the XGnPs-catalyzed ORR process, we performed the RDE measurements in an O2-saturated, 0.1 M KOH aqueous solution at various rotating speeds and a constant scan rate of 0.01 V s−1. As shown in Figures S11 and S12, the diffusion current densities increased with increasing the rotating rates, while the onset potentials remained almost constant. Once again, the limiting current densities for XGnPs are much higher than those for the pristine graphite at any constant rotation rate. It is well known that the number of electron transfers (n) per O2 molecule involved in the ORR process can be calculated from the slope of the Koutecky-Levich equation (see Experimental section in ESI)46,47. As shown in Figure S11f, the experimentally determined value (n = 4.0) at the limiting potential of −0.8 V for Pt/C agrees well with the theoretically calculated one (n = 4.0), indicating that a four-electron oxygen reduction process. The corresponding numbers of electrons transferred per O2 molecule at −0.8 V for the pristine graphite, ClGnP, BrGnP and IGnP were calculated to be 2.0, 3.5, 3.8 and 3.9 respectively (Figure S12f). The number of transferred electrons for the pristine graphite is close to the classical two-electron process, whereas the ORR processes at all the XGnP electrodes are much closer to an ideal four-electron process.
The origin of ORR activity enhancement with the nitrogen-doped carbon-based materials has been previously attributed to the higher electronegativity (χ) of the nitrogen (3.04) than carbon (2.55) to polarize adjacent carbon atoms in graphitic frameworks to efficiently facilitate the O2 adsorption and charge-transfer12. Considering the difference in the electronegativity between halogens (Cl = 3.16, Br = 2.96 and I = 2.66) and carbon (2.55), the reverse order ORR activities for ClGnP ≪ BrGnP < IGnP observed in this study seems to be contradicted to the doping-induced charge-transfer mechanism12. However, it is noted that the atomic sizes of Br and I are larger than that of Cl and hence the valence electrons of Br and I are much loosely bound than those of Cl for facilitating charge polarization in the BrGnP and IGnP electrodes. Unlike Cl, Br and I can form partially ionized bonds of –Br+– and –I+– (Figures S7d–S7f) to further enhance the charge-transfer because of their relatively large atomic sizes.
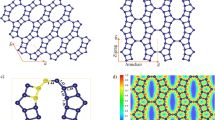
The above scenarios are supported by the first-principle DFT calculations for various types of edge structures of the XGnPs. As observed in XPS spectra (Figures S7c–7f), only two distinct types of edge configurations are considered; namely (i) the bond formations between halogen atoms and the sp2 carbons as dangling bonds at the zigzag edge (Figures S13a–S13c) and (ii) the substitutions of halogen atoms with the C sites (i.e., –Cl+–, –Br+– and –I+–) at the zigzag edge (Figures 4a–4c). When halogen atoms form the single sp2 C-X dangling bond, the edges does not have particular binding affinity with O2 molecule (Figures S13d–S13f). As a result of total energy minimization, the adsorption of O2 molecules is no more than the dispersive intermolecular physisorption. However, when halogen atoms substitute for the C sites in the zigzag edges as shown in Figures 4a–4c, the halogenated edges revealed decent binding affinity with O2. The PBE density functional calculations resulted in the binding of O2 with the strength of 0.71, 0.75 and 1.04 eV, respectively, for the cases of Cl (Figure 4a), Br (Figure 4b) and I (Figure 4c) with respect to the triplet state of free O2 in the vacuum. On the basis of the occupation of the lowest-unoccupied molecular orbital (LUMO) level of O2 as shown in Figures 4d–4f, it is noteworthy that the efficiency of the charge transfer between the halogen and adsorbed O2 follows the atomic size in the order of Cl < Br < I. Accordingly, the O-O bond length increases in that order (see Figures 4a–4c). The results suggest that the edges of halogenated graphene have enough activities to attract O2 and to weaken the O-O bond of the adsorbed O2, which is favorable for an efficient development into water upon reduction and protonation.

The optimized O2 adsorption geometries onto XGnPs, in which halogen covalently linked to two sp2 carbons.
(a) ClGnP; (b) BrGnP; (c) IGnP. The projected density of electronic states onto O2: (d) ClGnP; (e) BrGnP; (f) IGnP for the cases of corresponding (a), (b) and (c), respectively. In (a), (b) and (c), the O-O bond length and the shortest C-O bond are shown in angstroms.
To investigate the cycle stability of the XGnP electrocatalysts, we performed accelerated degradation test (ADT) by continuous chronoamperometric measurements at −0.25 V in an O2-saturated 0.1 M aq. KOH solution at a rotation rate of 1600 rpm. As shown in Figure 5a, the current densities for all of the electrodes initially decreased with time. However, the XGnP electrodes exhibited a relatively slow decay rate, maintaining 85.6–87.4% of their initial currents even after 10,000 s, in respect to the commercial Pt/C electrode (62.5%). These results indicate that the XGnP electrocatalysts have a much better electrochemical stability than the commercial Pt/C electrocatalyst. Furthermore, a sharp decrease in the ORR current density was observed for the commercial Pt/C electrocatalyst upon the additions of methanol while the XGnP electrodes and even the pristine graphite were insensitive to the methanol addition. For a methanol fuel cell, methanol crossover from anode to cathode could diminish cathodic performance through the depolarizing effect. Similar test has also been done to check the CO poisoning effect and the XGnP electrodes, along with the pristine graphite, were found to be free from the CO poisoning. In comparison with many precedent carbon-based ORR catalysts (Table S6), the preparation of XGnPs via ball-milling is very simple for practicality with overall better performance. Therefore, the above results unambiguously indicate that the XGnPs have much higher selectivity toward ORR than the commercial Pt/C electrocatalyst and are free from the CO poisoning, promising for applications in fuel cells and other energy-related devices.

(a) Current-time (i–t) chronoamperometric response of the pristine graphite, XGnPs and commercial Pt/C electrodes at −0.25 V vs. Ag/AgCl in O2-saturated 0.1 M aq. KOH solution at a rotation rate of 1600 rpm. Note that the blue and green curves are almost overlapped with each other. (b) The current-time (j–t) chronoamperometric responses for ORR at the pristine graphite, XGnPs and commercial Pt/C electrocatalysts in an O2-saturated 0.1 M aq. KOH solution at −0.40 V vs. Ag/AgCl and 3.0 M methanol or carbon monoxide (CO, 10 ppm/min) were added at around 360 s.
In summary, we have, for the first time, used a simple ball-milling method to synthesize edge-halogenated graphene nanoplatelets (XGnPs). The resultant XGnPs were tested as cathode electrodes of fuel cells and revealed remarkable electrocatalytic activities for ORR with high tolerance to methanol crossover/CO poisoning effects and longer-term stability than those of the pristine graphite and commercial Pt/C electrocatalysts. To understand the underlying mechanism, first-principle DFT calculations were performed for various edge configurations. We found that the edges of halogenated graphene have favorable binding affinity with O2 molecule and the O-O bond strengths are weakened as a result of the halogenation-induced charge transfer. Our results suggest new insights and practical methods for designing edge-functionalized GnPs as high-performance metal-free ORR electrocatalysts through low-cost and scalable ball-milling techniques.
Methods
Syntheses of XGnPs
XGnPs were prepared simply by ball-milling the pristine graphite in a planetary ball-mill capsule in the presence of Cl2, Br2 and I2, respectively. In a typical experiment, the pristine graphite (5.0 g) was placed into a stainless steel capsule containing stainless steel balls (500 g, diameter 5 mm). The capsule was then sealed and degassed by five cycles of charging and discharging argon after applying reduced pressure (0.05 mmHg). Thereafter, Cl2 gas was charged through a gas inlet with cylinder pressure of 8.75 atm. The capsule was then fixed in the planetary ball-mill machine and agitated with 500 rpm for 48 h. The resultant product was Soxhlet extracted with methanol to get rid of small molar mass organic impurities and 1 M aq. HCl solution to remove metallic impurities, if any. Final product was then freeze-dried at −120°C under a reduced pressure (0.05 mmHg) for 48 h to yield 6.09 g (ClGnP has at least 1.09 g of Cl uptake) of dark black ClGnP powder. BrGnP (6.93 g, 1.93 g of Br uptake) and IGnP (6.86 g, 1.86 g of I uptake) samples were also prepared following the similar ball-milling and work-up procedures.
References
Stamenkovic, V. R. et al. Improved oxygen reduction activity on Pt3Ni (111) via increased surface site availability. Science 315, 493–497 (2007).
Zhang, J. et al. Mixed-metal Pt monolayer electrocatalysts for enhanced oxygen reduction kinetics. J. Am. Chem. Soc. 127, 12480–12481 (2005).
Xiao, L., Zhuang, L., Liu, Y. & Lu, J. Activating Pd by morphology tailoring for oxygen reduction. J. Am. Chem. Soc. 131, 602–608 (2008).
Peng, Z. & Yang, H. Synthesis and oxygen reduction electrocatalytic property of Pt-on-Pd bimetallic heteronanostructures. J. Am. Chem. Soc. 131, 7542–7543 (2009).
Zhang, S., Shao, Y., Yin, G. & Lin, Y. Carbon nanotubes decorated with Pt nanoparticles via electrostatic self-assembly: a highly active oxygen reduction electrocatalyst. J. Mater. Chem. 20, 2826–2830 (2010).
Lim, B. et al. Pd-Pt bimetallic nanodendrites with high activity for oxygen reduction. Science 324, 1302–1305 (2009).
Zhang, S., Shao, Y., Yin, G. & Lin, Y. Self-assembly of Pt nanoparticles on highly graphitized carbon nanotubes as an excellent oxygen-reduction catalyst. Appl. Catal. B-Envion. 102, 372–377 (2011).
Huang, S. Y., Ganesan, P., Park, S. & Popov, B. N. Development of a titanium dioxide-supported platinum catalyst with ultrahigh stability for polymer electrolyte membrane fuel cell applications. J. Am. Chem. Soc. 131, 13898–13899 (2009).
Wu, G., More, K. L., Johnston, C. M. & Zelenay, P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron and cobalt. Science 332, 443–447 (2011).
Lefèvre, M., Proietti, E., Jaouen, F. & Dodelet, J. P. Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009).
Byon, H. R., Suntivich, J. & Shao-Horn, Y. Graphene-based non-noble-metal catalysts for oxygen reduction reaction in acid. Chem. Mater. 23, 3421–3428 (2011).
Gong, K., Du, F., Xia, Z., Durstock, M. & Dai, L. Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323, 760–764 (2009).
Wang, S., Yu, D. & Dai, L. Polyelectrolyte functionalized carbon nanotubes as efficient metal-free electrocatalysts for oxygen reduction. J. Am. Chem. Soc. 133, 5182–5185 (2011).
Yang, L. et al. Boron-doped carbon nanotubes as metal-free electrocatalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 123, 7270–7273 (2011).
Sheng, Z. H. et al. Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis. ACS Nano 5, 4350–4358 (2011).
Tang, Y., Allen, B. L., Kauffman, D. R. & Star, A. Electrocatalytic activity of nitrogen-doped carbon nanotube cups. J. Am. Chem. Soc. 131, 13200–13201 (2009).
Shanmugam, S. & Osaka, T. Efficient electrocatalytic oxygen reduction over metal free-nitrogen doped carbon nanocapsules. Chem. Commun. 47, 4463–4465 (2011).
Sheng, Z. H., Gao, H. L., Bao, W. J., Wang, F. B. & Xia, X. H. Synthesis of boron doped graphene for oxygen reduction reaction in fuel cells. J. Mater. Chem. 22, 390–395 (2011).
Yao, Z. et al. Catalyst-free synthesis of iodine-doped graphene via a facile thermal annealing process and its use for electrocatalytic oxygen reduction in an alkaline medium. Chem. Commun. 48, 1027–1029 (2012).
Zheng, J. et al. Production of graphite chloride and bromide using microwave sparks. Sci. Rep. 2, 662 (2012).
Liu, R., Wu, D., Feng, X. & Müllen, K. Nitrogen-doped ordered mesoporous graphitic arrays with high electrocatalytic activity for oxygen reduction. Angew. Chem. Int. Ed. 122, 2619–2623 (2010).
Yu, D., Zhang, Q. & Dai, L. Highly efficient metal-free growth of nitrogen-doped single-walled carbon nanotubes on plasma-etched substrates for oxygen reduction. J. Am. Chem. Soc. 132, 15127–15129 (2010).
Zheng, Y. et al. Nanoporous graphitic-C3N4@carbon metal-free electrocatalysts for highly efficient oxygen reduction. J. Am. Chem. Soc. 133, 20116–20119 (2011).
Jeon, I. Y. et al. Formation of large-area nitrogen-doped graphene film prepared from simple solution casting of edge-selectively functionalized graphite and its electrocatalytic activity. Chem. Mater. 23, 3987–3992 (2011).
Qu, L., Liu, Y., Baek, J. B. & Dai, L. Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 4, 1321–1326 (2010).
Liu, Z. W. et al. Phosphorus-doped graphite layers with high electrocatalytic activity for the O2 reduction in an alkaline medium. Angew. Chem. Int. Ed. 123, 3315–3319 (2011).
Yang, Z. et al. Sulfur-doped graphene as an efficient metal-free cathode catalyst for oxygen reduction. ACS Nano 6, 205–211 (2011).
Yang, S. et al. Efficient synthesis of heteroatom (N or S)-doped graphene based on ultrathin graphene oxide-porous silica sheets for oxygen reduction reactions. Adv. Funct. Mater 22, 3634–3640 (2012).
Wang, S. et al. Vertically aligned BCN nanotubes as efficient metal-free electrocatalysts for the oxygen reduction reaction: a synergetic effect by co-doping with boron and nitrogen. Angew. Chem. Int. Ed. 50, 11756–11760 (2011).
Wang, S. et al. BCN graphene as efficient metal-free electrocatalyst for the oxygen reduction reaction. Angew. Chem. Int. Ed. 51, 4209–4212 (2012).
Zhang, M. & Dai, L. Carbon nanomaterials as metal-free catalysts in next generation fuel cells. Nano Energy 1, 514–517 (2012).
Dai, L., Chang, D. W., Baek, J.-B. & Lu, W. “Carbon nanomaterials for advanced energy conversion and storage”. Small 8, 1130–1166 (2012).
Jeon, I.-Y. et al. Edge-carboxylated graphene nanosheets via ball milling. Proc. Natl. Acad. Sci. USA 109, 5588–5593 (2012).
Jaccaud, M., Faron, R., Devilliers, D. & Romano, R. Fluorine. Ullmann's Encyclopedia of Industrial Chemistry (John Wiley and Sons, Inc., New York, 2000).
Moore, J. W., Stanitski, C. L. & Jurs, P. C. Principles of chemistry: the molecular science. (Brooks/Cole Publishing Company, 2009).
Cho, D. W. et al. Nature and kinetic analysis of carbon-carbon bond fragmentation reactions of cation radicals derived from SET-oxidation of lignin model compounds. J. Org. Chem. 75, 6549–6562 (2010).
Li, Z., Lu, C., Xia, Z., Zhou, Y. & Luo, Z. X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 45, 1686–1695 (2007).
Dikin, D. A. et al. Preparation and characterization of graphene oxide paper. Nature 448, 457–460 (2007).
Zhu, Y. et al. Graphene and graphene oxide: synthesis, properties and applications. Adv. Mater. 22, 3906–3924 (2010).
Everett, D. H. & Chemistry, R. S. Basic principles of colloid science vol. 144 (Royal Society of Chemistry, London, 1988).
Becerril, H. A. et al. Evaluation of solution-processed reduced graphene oxide films as transparent conductors. ACS Nano 2, 463–470 (2008).
Stankovich, S. et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45, 1558–1565 (2007).
Zheng, Q. B. et al. Improved electrical and optical characteristics of transparent graphene thin films produced by acid and doping treatments. Carbon 49, 2905–2916 (2011).
Jung, Y., Hwang, S. J. & Kim, S. J. Spectroscopic evidence on weak electron transfer from intercalated iodine molecules to single-walled carbon nanotubes. J. Phys. Chem. C 111, 10181–10184 (2007).
Jeon, I.-Y. et al. Large-scale production of edge-selectively functionalized graphene nanoplatelets via ball milling and their use as metal-free electrocatalysts for oxygen reduction reaction. J. Am. Chem. Soc. 135, 1386–1393 (2012).
Jung, H. Y., Park, S. & Popov, B. N. Electrochemical studies of an unsupported PtIr electrocatalyst as a bifunctional oxygen electrode in a unitized regenerative fuel cell. J. Power Sources 191, 357–361 (2009).
Zhang, S., Shao, Y., Yin, G. & Lin, Y. Stabilization of platinum nanoparticle electrocatalysts for oxygen reduction using poly (diallyldimethylammonium chloride). J. Mater. Chem. 19, 7995–8001 (2009).
Acknowledgements
This research was supported by World Class University (WCU), US-Korea NBIT, Mid-Career Researcher (MCR), Converging Research Center (CRC) and Basic Research Laboratory (BRL) programs through the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Science and Technology (MEST), US Air Force Office of Scientific Research through Asian Office of Aerospace R&D (AFOSR-AOARD) and AFOSR (FA9550-12-1-0037, FA-9550-12-1-0069).
Author information
Authors and Affiliations
Contributions
J.B.B. conceived the reaction system and oversaw all the research phases. I.-Y.J., H.-J.C., M.C. conducted experiments. J.M.S., S.M.J., M.J.K. and S.Z. carried out the characterizations. Z.X. and N.P. were involved in the Ab Initio study of the new material by DFT. J.-B.B., N.P. and L.D. wrote the paper and discussed the results. All authors contributed and commented on this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Supporting Video
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Jeon, IY., Choi, HJ., Choi, M. et al. Facile, scalable synthesis of edge-halogenated graphene nanoplatelets as efficient metal-free eletrocatalysts for oxygen reduction reaction. Sci Rep 3, 1810 (2013). https://doi.org/10.1038/srep01810
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01810
This article is cited by
-
Atomic design of carbon-based dual-metal site catalysts for energy applications
Nano Research (2023)
-
Rational design of carbon-based electrocatalysts for enhancing redox reactions in rechargeable metal batteries
Nano Research (2023)
-
Lignin derived carbon materials: current status and future trends
Carbon Research (2022)
-
Preparation of Pt electrocatalyst supported by novel, Ti(1−x)MoxO2-C type of composites containing multi-layer graphene
Reaction Kinetics, Mechanisms and Catalysis (2022)
-
Derived from Diaryl-λ3-Iodane-Containing Polyoxometalate: Iodine-Doped Molybdenum Carbide for Efficient Electrocatalytic Hydrogen Evolution
Journal of Cluster Science (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
